UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For
the fiscal year ended
OR
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission
File Number:
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of | (IRS Employer | |
| incorporation or organization) | Identification No.) |
(Address of Principal Executive Offices)(Zip Code)
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Trading Symbol | Name of Each Exchange on Which Registered | ||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| Accelerated filer ☐ | |
| Non-accelerated filer ☐ | Smaller
reporting company |
| Emerging
growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes
☐ No
As
of June 30, 2025, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market
value of the common stock held by non-affiliates of the registrant was approximately $
As of February 25, 2026, there were shares of the registrant’s common stock outstanding.
TABLE OF CONTENTS
| 2 |
As used in this Annual Report on Form 10-K (this “Annual Report”), unless indicated or the context requires otherwise, the terms the “Company,” “Harrow,” “we,” “us” and “our” refer to Harrow, Inc. and its consolidated subsidiaries.
In addition to historical information, the following discussion contains forward-looking statements regarding future events and our future performance. In some cases, you can identify forward-looking statements by terminology such as “will,” “may,” “should,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “forecasts,” “potential” or “continue” or the negative of these terms or other comparable terminology. All statements made in this Annual Report other than statements of historical fact are forward-looking statements. These forward-looking statements involve risks and uncertainties and reflect only our current views, expectations and assumptions with respect to future events and our future performance. If risks or uncertainties materialize or assumptions prove incorrect, actual results or events could differ materially from those expressed or implied by such forward-looking statements. Risks that could cause actual results to differ from those expressed or implied by the forward-looking statements we make include, among others, risks related to: liquidity or results of operations; our ability to successfully implement our business plan, manage our pharmacy operations, service our debt, obtain financing necessary to operate our business, recruit and retain qualified personnel, manage any growth we may experience and successfully realize the benefits of our previous acquisitions and any other acquisitions and collaborative arrangements we may pursue; the ongoing communications with the U.S. Food and Drug Administration relating to compliance and quality plans at our outsourcing facility in New Jersey; competition from pharmaceutical companies, outsourcing facilities and pharmacies; general economic and business conditions, including inflation and supply chain challenges; regulatory and legal risks and uncertainties related to our pharmacy operations and the pharmacy and pharmaceutical business in general; physician interest in and market acceptance of our current and any future products and formulations and compounding pharmacies generally; our limited operating history; and the other risks and uncertainties described under the heading “Risk Factors” in Part I, Item 1A of this Annual Report. You should not place undue reliance on forward-looking statements. Forward-looking statements speak only as of the date they are made and, except as required by law, we undertake no obligation to revise or publicly update any forward-looking statement for any reason.
We have registered trademarks, copyrights and/or pending trademark and copyright applications for a number of proprietary names in the United States of America (“U.S.”), including, but not limited to: VEVYE®, IHEEZO®, ILEVRO®, TRIESENCE®, ImprimisRx®, and LessDrops®. We may choose to pursue trademark protection in other jurisdictions for one or more of these or other marks in the future. All other trademarks, service marks and trade names included or incorporated by reference into this Annual Report, are the property of their respective owners.
| 3 |
PART I
ITEM 1. BUSINESS
Overview
We are a leading provider of ophthalmic disease management solutions in North America, and were founded with a commitment to deliver safe, effective, accessible, and affordable medications that enhance patient compliance and improve clinical outcomes. For over a decade, we have partnered with U.S. eyecare professionals to develop a comprehensive portfolio of high-quality products used to manage ophthalmic conditions affecting both the front and back of the eye, such as dry eye disease, wet (or neovascular) age-related macular degeneration, cataracts, refractive errors, glaucoma, and a range of other ocular surface conditions and retina diseases. By prioritizing clinical value – to the provider and the patient – Harrow empowers professionals to enhance patient outcomes and preserve vision. By combining our culture of creativity, entrepreneurship and groundbreaking innovation with operational discipline and strong financial performance, we are building a future where life-changing ophthalmic treatments are within reach for all.
Branded Ophthalmic Pharmaceuticals
Over the past several years, we have expanded our portfolio of the U.S. Food and Drug Administration (the “FDA”)-approved ophthalmic products through acquisitions, licensing transactions, and internal investment. These efforts are focused primarily on the U.S. and Canadian markets. We believe continued investment in our branded portfolio supports our ability to offer eyecare prescribers and patients access to a broader range of ophthalmic therapies across multiple disease states. We own U.S. commercial rights to the following products, which we market and sell:
| ● | IHEEZO® (chloroprocaine hydrochloride ophthalmic gel) 3%, a low-viscosity gel indicated for ocular surface anesthesia. |
| ● | VEVYE® (cyclosporine ophthalmic solution) 0.1%, utilizes a novel water-free vehicle (perfluorobutylpentane) based on semifluorinated alkanes, indicated for the treatment of the signs and symptoms associated with dry eye disease. |
| ● | TRIESENCE® (triamcinolone acetonide injectable suspension) 40 mg/ml, a steroid injection for the treatment of certain ophthalmic diseases and for visualization during vitrectomy. |
| ● | BYOOVIZ® (ranibizumab-nuna) 0.05mL injection, the first FDA-approved LUCENTIS biosimilar indicated for the treatment of patients with Neovascular (Wet) Age-Related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), and Myopic Choroidal Neovascularization (mCNV). We expect to commercially launch BYOOVIZ in mid-2026. |
| ● | OPUVIZ® (aflibercept-yszy) 0.05mL injection, an FDA-approved EYLEA biosimilar indicated for the treatment of patients with Wet AMD, Macular Edema following RVO, Diabetic Macular Edema (DME), and Diabetic Retinopathy (DR). We expect to commercially launch OPUVIZ in mid-2027. |
| ● | BYQLOVITM (clobetasol propionate ophthalmic suspension) 0.05% a high-potency ophthalmic corticosteroid formulated using proprietary APNT® nanoparticle formulation technology, indicated for the treatment of post-operative inflammation and pain following ocular surgery. We expect to commercially launch BYQLOVI in mid-2026. |
| ● | VIGAMOX® (moxifloxacin hydrochloride ophthalmic solution) 0.5%, a fluoroquinolone antibiotic eye drop for the treatment of bacterial conjunctivitis caused by susceptible strains of organisms. |
| 4 |
| ● | ILEVRO® (nepafenac ophthalmic suspension) 0.3%, a non-steroidal, anti-inflammatory eye drop indicated for pain and inflammation associated with cataract surgery. |
| ● | FLAREX® (fluorometholone acetate ophthalmic suspension) 0.1%, a corticosteroid prepared as a sterile topical ophthalmic suspension indicated for use in the treatment of steroid-responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the eye. |
| ● | NATACYN® (natamycin ophthalmic suspension) 5%, a sterile, antifungal drug for the treatment of fungal blepharitis, conjunctivitis, and keratitis caused by susceptible organisms, including Fusarium solani keratitis. |
| ● | TOBRADEX® ST (tobramycin and dexamethasone ophthalmic suspension) 0.3%/0.05%, a topical antibiotic and corticosteroid combination for steroid-responsive inflammatory ocular conditions for which a corticosteroid is indicated and where superficial bacterial ocular infection or a risk of bacterial ocular infection exists. |
| ● | ZERVIATE® (cetirizine ophthalmic solution) 0.24%, a histamine-1 (H1) receptor antagonist indicated for treatment of ocular itching associated with allergic conjunctivitis. |
| ● | VERKAZIA® (cyclosporine ophthalmic emulsion) 0.1%, an orphan designated drug that is a calcineurin inhibitor immunosuppressant indicated for the treatment of vernal keratoconjunctivitis. |
| ● | NEVANAC® (nepafenac ophthalmic suspension) 0.1%, a non-steroidal, anti-inflammatory eye drop indicated for pain and inflammation associated with cataract surgery. |
| ● | FRESHKOTE® Preservative Free (PF) is a lubricant eye drop that does not require a prescription and temporarily relieves burning, itching and other dry eye symptoms. |
| ● | MAXITROL® (neomycin and polymyxin B sulfates and dexamethasone ophthalmic suspension) is an eye drop used to treat steroid-responsive inflammatory ocular conditions where bacterial infection or a risk of bacterial ocular infection exist. |
| ● | MAXIDEX® (dexamethasone ophthalmic suspension) 0.1%, a steroid eye drop for steroid-responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the globe. |
| ● | IOPIDINE® 1% (apraclonidine hydrochloride), an ophthalmic solution in a sterile isotonic solution indicated to control or prevent post-surgical elevations in intraocular pressure that occur in patients after argon laser trabeculoplasty, argon laser iridotomy or Nd:YAG posterior capsulotomy. |
| ● | IOPIDINE® 0.5% (apraclonidine hydrochloride) an ophthalmic solution indicated for short-term adjunctive therapy in patients on maximally tolerated medical therapy who require additional intraocular pressure (or IOP) reduction. |
| 5 |
We also own U.S. rights to some discontinued products (MOXEZA, VEXOL, ECONOPRED and TOBRASONE). In February 2024, we announced that we out-licensed Canadian rights for VERKAZIA, Cationorm® PLUS (a preservative-free formulation for dry eye or allergy relief), VEVYE, ZERVIATE and IHEEZO to Apotex Inc. (“Apotex”). We also own worldwide rights to NATACYN and FRESHKOTE.
R&D Development Pipeline
Our development pipeline is focused on developing and commercializing differentiated pharmaceutical therapies designed to address unmet needs in eye care and selected adjacent markets. The pipeline is weighted toward late-stage and near-commercial assets with demonstrated clinical utility and clearer regulatory pathways, and we seek to advance programs through a disciplined, capital-efficient development strategy. Current development-stage programs include:
| ● | MELT-300 (ketamine + midazolam ODT): completed Phase 3 clinical program; potential launch in 2028. MELT-300 is a patented, sublingual/orally disintegrating tablet that combines a fixed dose of midazolam (3 mg) and ketamine (50 mg) and is designed to provide rapid, predictable sedation without IV administration, including in cataract and other outpatient procedures. |
| ● | H-N08 (triamcinolone acetonide): current work is focused on chemistry, manufacturing and controls (CMC) optimization, and is expected to move into the clinic before the end of 2026, with a potential launch in 2028. H-N08 is an ophthalmic program built around triamcinolone acetonide intended to support inflammatory indications such as uveitis and/or improve intraoperative visualization in posterior-segment surgery (e.g., during vitrectomy). |
| ● | CR-01 (conjunctival delivery device): is in the proof-of-concept evaluation stage, with potential launch as early as 2029. CR-01 is a device-enabled program intended to deliver therapy via the conjunctiva for ocular neoplasia, a rare disease. |
| ● | MELT-210 (midazolam ODT): is currently in clinical development stage; with a potential launch in 2028. MELT-210 is a sublingual/orally disintegrating tablet being developed as a needle-free option for procedural sedation and related anxiolysis/amnesia needs, and has also been used in MELT-300’s clinical program as a midazolam-only comparator. |
Development timelines and potential launch timing are subject to change based on clinical results, regulatory feedback, manufacturing readiness, and other factors.
MELT-300
We believe MELT-300 represents a transformative opportunity, building on more than a decade of real-world experience with MKO Melt® — a compounded sublingual sedation product sold by ImprimisRx and currently administered by over 800 U.S. ophthalmic institutions, primarily for used for procedural sedation during cataract surgery. As a potential FDA-approved successor, MELT-300 is a patented, sublingually delivered formulation of a fixed dose of midazolam (3mg) and ketamine (50mg) designed to provide rapid, predictable sedation without the need for intravenous administration. The MELT-300 Phase 2 and Phase 3 clinical programs previously demonstrated statistical superiority to midazolam alone. The innovative approach to sedation MELT-300 offers has the potential to transform patient experiences across a wide range of office-based and outpatient procedures, addressing the healthcare system’s growing demand to reduce exposure to opioids, including fentanyl.
The MELT-300 program was the subject of a Special Protocol Assessment (SPA) with the FDA, confirming that the completed Phase 3 study design, statistical approach, and endpoints adequately support a future regulatory submission. Having completed the Phase 3 program, our focus now turns to advancing MELT-300 toward FDA approval and commercialization.
In support of a new drug application (an “NDA”) filing, we recently initiated one non-clinical animal study and three pharmacokinetic (“PK”) studies to generate the balance of the data we believe is necessary for an NDA package. Following completion of these studies, we expect to prepare and submit an NDA for MELT-300 in the first half of 2027. If promptly approved by the FDA, we expect to commercially launch MELT-300 in the second half of 2028.
| 6 |
We believe these next steps position MELT-300 to become the first FDA-approved, non-opioid, non-IV sublingual sedation therapy in the U.S., representing a meaningful growth opportunity for Harrow and a major advancement in patient-centric procedural care. With patent coverage in the U.S. and other international markets and potential applications beyond ophthalmology—including gastroenterology, dental care, and other outpatient settings where sedation or anxiety management may be beneficial, such as diagnostic imaging, endoscopy and pre-anesthesia—MELT-300 may provide us the opportunity to expand into procedural sedation and anxiety management indications outside of eye care domestically and in international markets.
ImprimisRx
ImprimisRx is our ophthalmology-focused pharmaceutical compounding business. Since inception in 2014, ImprimisRx has provided ophthalmologists, optometrists, and their patients access to compounded medications intended to address needs that may be unmet by commercially available products, including combination therapies, alternative dosage strengths, and preservative-free formulations. Depending on formulation, applicable state requirements, and patient need, ImprimisRx products may be dispensed as patient-specific prescriptions from our 503A pharmacy or manufactured for in-office use in our FDA-registered 503B outsourcing facility in New Jersey. Our current ophthalmology formulary includes over 30 compounded formulations, many of which are patented or patent-pending, and our customer base includes more than 10,000 U.S. eyecare-dedicated prescribers and institutions.
We operate two compounding facilities in Ledgewood, New Jersey. One facility is registered with the FDA as an outsourcing facility under Section 503B of the Federal Food, Drug and Cosmetic Act (the “FDCA”) (NJOF). The other facility is a licensed pharmacy operating under Section 503A of the FDCA (RxNJ). All compounded products we sell, produce, and dispense are made in the United States. We believe our current infrastructure supports continued scaling within the current regulatory landscape, and we may pursue additional capacity, redundancy, and market access through investments, partnerships, or strategic transactions.
Pharmaceutical Compounding
Pharmaceutical compounding involves preparing customized formulations for patients when commercially available products do not meet a patient’s clinical needs. Compounded formulations contain FDA-approved ingredients, but the compounded formulations themselves are not FDA-approved. Compounding is subject to extensive federal and state regulation and oversight, which can affect permissible activities, cost structure, and the ability to dispense into particular states.
Carved-Out Subsidiaries (De-Consolidated Businesses)
We have an ownership interest in Surface Ophthalmics, Inc. (“Surface”) and hold royalty interests in some of Surface’s drug candidates. Surface is pursuing market approval for its drug candidates under the FDCA, including in some instances under the abbreviated pathway described in Section 505(b)(2), which permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. We previously held ownership interests in Eton Pharmaceuticals, Inc. (“Eton”) and sold the last of our interests in April 2024.
Acquisition of the Remaining Equity Interests of Melt Pharmaceuticals
Prior to November 2025, we held a minority ownership interest in Melt Pharmaceuticals, Inc. (“Melt”), a clinical-stage pharmaceutical company focused on the development and commercialization of proprietary non-intravenous, sedation and anesthesia therapeutics for human medical procedures in hospital, outpatient, and in-office settings. Melt sought regulatory approval for its proprietary technologies, where possible.
As of December 31, 2024, we owned approximately 45% of Melt’s equity and voting interests issued and outstanding, along with a mid-single digit royalty on future net sales of MELT-300.
| 7 |
In September 2025, we entered into an Agreement and Plan of Merger (the “Merger Agreement”) by and among Harrow, Harrow Acquisition Sub, Inc., a wholly owned subsidiary of Harrow, Melt, and D. Brad Osborne, as stockholder representative. Under the terms of the Merger Agreement and a related milestone payment agreement, we agreed to acquire the remaining equity interests of Melt in exchange for an initial cash payment of approximately $4,300,000 at closing, and contingent consideration consisting of cash and Harrow equity upon achievement of (i) FDA approval of the MELT-300 product candidate, (ii) coding and reimbursement of the MELT-300 product candidate, and (iii) various one-time sales milestones. The regulatory and commercial milestones must be achieved on or before December 31, 2035.
The Melt acquisition closed on November 17, 2025, and was treated as an asset acquisition for accounting purposes. As a result of such transaction, Melt’s drug candidates are now owned by Harrow and its research and development (“R&D”) activities subsequent to the acquisition are included in Harrow’s consolidated financial results as of the year ended December 31, 2025.
Sales and Marketing
The focus of our sales and marketing is in the U.S. We do, however, believe that our drug candidates and drug products could have commercial appeal in international markets, and have engaged distributors and entered into out-licensing arrangements for certain of our products and proprietary formulations in certain non-U.S. markets, including Canada. Our sales and marketing activities consist primarily of efforts to educate doctors, ambulatory surgery centers, healthcare systems, hospitals and other users throughout the U.S. about our drug products. We expect that we may experience growth in the sales of our products in future periods, particularly in light of our recent product launches and commercial campaigns. However, we may not be successful in doing so, whether due to the size of the markets for such products, which could be smaller than we expect, the timing of market entry relative to competitive products, the availability of alternative compounded formulations or FDA-approved drugs, the price of our products relative to alternative products or the success of our sales and marketing efforts, which is dependent on our ability to further build and continue to grow a qualified and adequate internal sales function.
We expect to continue to acquire and/or develop additional FDA-approved products that allow us to leverage our existing commercial infrastructure to promote, sell, and ultimately bring these products to market. As we execute this strategy, we will continue to expand our sales and marketing team, expertise and expenses.
Supply Chains
100% of our ImprimisRx finished compounded products are made in the U.S. at our compounding facilities located in New Jersey.
We do not manufacture any of our branded pharmaceutical products and rely on third party manufacturing partners to make finished goods. The following table describes by product the country where our finished branded products are made. In some instances, multiple countries are listed to reflect either (i) expected changes in our contract manufacturer and location; and (ii) in certain cases, to reflect the country of a second contract manufacturer site:
| Product | Country Finished Product Is Manufactured | |
| IHEEZO | France | |
| VEVYE | U.S. and Spain | |
| TRIESENCE | U.S. | |
| VIGAMOX | Belgium | |
| ILEVRO | Belgium | |
| FLAREX | U.S., by end of 2026 production is expected to be in Taiwan | |
| NATACYN | U.S. | |
| TOBRADEX ST | U.S.; by end of 2026 production is expected to be in Taiwan | |
| ZERVIATE | France; by end of 2026 production is expected to be in Spain | |
| VERKAZIA | France | |
| NEVANAC | U.S.; by end of 2026 production is expected to be in Belgium | |
| FRESHKOTE | France; by end of 2026 production is expected to be in Spain | |
| MAXIDEX | U.S.; by end of 2026 production is expected to be in Belgium | |
| MAXITROL | Belgium | |
| IOPIDINE 1% | France | |
| BYQLOVI | Taiwan |
| 8 |
Ophthalmology Market
Ophthalmic pharmaceuticals are used across several major categories of eye care, including high-volume procedures (such as cataract/lens procedures and refractive surgeries like LASIK), chronic ocular surface conditions (including dry eye disease), retina disease management (often involving physician-administered therapies delivered by intravitreal injection), and posterior-segment surgical care (including vitrectomy and related visualization and inflammation management). These settings drive demand for different classes of ophthalmic products—such as anesthetics, anti-infectives, anti-inflammatories (including corticosteroids and NSAIDs), immunomodulators, and ocular surface lubricants—used before, during, and after procedures and for ongoing disease management. Utilization across these categories is influenced by procedure volumes and clinical practice patterns, the availability of therapeutic alternatives (including generics and biosimilars), and market access factors such as formulary placement, prior authorization/step therapy, reimbursement, and patient out-of-pocket costs. Our portfolio is focused on these major ophthalmic categories and spans products used in ocular surface disease, perioperative care, posterior-segment surgical and retina settings. We have limited exposure to glaucoma therapies.
Competition
The pharmaceutical industry is highly competitive. We compete with branded and generic pharmaceutical companies, biosimilar manufacturers, and other companies developing or commercializing ophthalmic therapies, including products used in ocular surface disease, perioperative care, and retina. Certain competitors have substantially greater financial, technical, manufacturing, and commercial resources than we do, and may be able to develop, obtain regulatory approval for, manufacture, market, and sell products more effectively than we can, including through larger sales forces, broader distribution networks, and greater access to capital. As a result, we may face competitive disadvantages in gaining or maintaining market share, achieving favorable formulary placement and reimbursement, securing manufacturing capacity and supply chain reliability, and sustaining pricing and margins.
Biotechnology and pharmaceutical technologies are subject to rapid and significant change. Our success depends in part on our ability to maintain a competitive position with respect to new therapies, delivery methods, and competitive entrants, including generics and biosimilars. Competitors may introduce products that are safer, more effective, easier to use, more durable, more convenient, or more cost-effective than our products, or that achieve greater market access through reimbursement or contracting advantages. New competitive products could reduce demand for, or the market opportunity of, our existing products and could render our product candidates or lifecycle management initiatives less attractive or commercially viable before we recover development or commercialization investments.
Competition also depends on factors such as clinical performance, physician adoption and practice patterns, timing of market entry, regulatory developments, product availability and supply reliability, pricing and patient affordability, third-party reimbursement, and the effectiveness of sales, marketing, and distribution efforts. If we are unable to compete effectively with current or future products of our competitors, our revenues, profitability, and growth prospects could be materially and adversely affected.
To the extent we offer compounded formulations through our pharmacy operations, those products also face competition from FDA-approved alternatives and other compounded products, and utilization may be affected by physician and patient preferences and applicable regulatory requirements.
| 9 |
Factors Affecting Our Performance
We believe the primary factors affecting our performance are our ability to increase revenues of our ophthalmic products, grow and gain operating efficiencies in our pharmacy operations, successfully adjust our operations to account for any future regulatory-related restrictions, optimize pricing and obtain reimbursement options for our ophthalmic products, and continue to pursue development and commercialization opportunities for certain of our ophthalmology and other assets that we have not yet made commercially available or have been recently launched. We believe we have built a tangible and intangible infrastructure that will allow us to scale revenues efficiently in the near and long-term. All of these activities will require increased costs and other resources, which we may not have or be able to obtain from operations or other sources. See Item 7. “Management’s Discussion and Analysis of Financial Condition and Result of Operations - Liquidity and Capital Resources.”
Medicare, Medicaid and Other Reimbursement Options
Sales in the U.S. of our marketed products are dependent, in large part, on the availability and extent of reimbursement from third-party payors, including private payor healthcare and insurance programs, health maintenance organizations, pharmacy benefit management companies, and government programs such as Medicare and Medicaid. See Item 1A. “Risk Factors” for risks related to reimbursement and government programs.
We participate in, and have certain price reporting obligations to, the Medicaid Drug Rebate program, state Medicaid supplemental rebate program(s), and other governmental pricing programs. We also have obligations to report the average sales price for certain drugs to the Medicare program. Under the Medicaid Drug Rebate program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available for our drugs under Medicaid and Part B of the Medicare program.
Medicare is a federal program that is administered by the federal government that covers individuals age 65 and over or that are disabled as well as those with certain health conditions. Medicare Part B generally covers drugs that must be administered by physicians or other health care practitioners; are provided in connection with certain durable medical equipment; or are certain oral anti-cancer drugs and certain oral immunosuppressive drugs. Medicare Part B pays for such drugs under a payment methodology based on the average sales price of the drugs. Manufacturers, including us, are required to report average sales price information to the Centers for Medicare & Medicaid Services (“CMS”) on a quarterly basis. The manufacturer-submitted information may be used by CMS to calculate Medicare payment rates. Starting in 2023, manufacturers are now required to pay refunds to Medicare for single-source drugs or biological products, or biosimilar biological products, reimbursed under Medicare Part B and packaged in single-dose containers or single-use packages for units of discarded drug reimbursed by Medicare Part B in excess of 10% of total allowed charges under Medicare Part B for that drug. Manufacturers that fail to pay refunds could be subject to civil monetary penalties. Further, starting in 2023, the Inflation Reduction Act of 2022 (“IRA”) established a Medicare Part B inflation rebate scheme, effective in 2023, under which, generally speaking, manufacturers will owe rebates if the average sales price of a Part B drug increases faster than the pace of inflation. Failure to timely pay a Part B inflation rebate is subject to a civil monetary penalty.
The IRA also created a drug price negotiation program under which, after being on the market for a certain period of time, the prices for certain high Medicare spending drugs and biological products provided to Medicare patients without generic or biosimilar competition will be capped by reference to, among other things, a specified non-federal average manufacturer price, starting in 2026. Failure to comply with requirements under the drug price negotiation program is subject to an excise tax and a civil monetary penalty. This or any other legislative change could impact the market conditions for our products.
| 10 |
BYOOVIZ, OPUVIZ, IHEEZO and TRIESENCE are covered under Medicare Part B and we are developing other product candidates and may acquire drug products that are also covered under Medicare Part B. In February 2023, we announced that CMS had issued a permanent, product specific J-code for IHEEZO (J2403) which became effective under the Healthcare Procedure Coding System (HCPCS) on April 1, 2023. TRIESENCE has a permanent product specific J-code (J3300) as well, which physicians can use for reimbursement purposes of that product. Similarly, BYOOVIZ and OPUVIZ have permanent product specific reimbursement codes of Q5124 and Q5153, respectively, which healthcare professionals can use for reimbursement of those biosimilar products.
New drugs approved by the FDA that are used in surgeries performed in a hospital outpatient departments or ambulatory surgical centers may receive a transitional pass-through reimbursement under Medicare, provided they meet certain criteria, including a “not insignificant” cost criterion. Pass-through status allows for separate payment (i.e., outside the packaged payment rate for the surgical procedure) under Medicare Part B, which consists of Medicare reimbursement for a drug based on a defined formula for calculating the minimum fee that a manufacturer may charge for the drug. Under current regulations of CMS, pass-through status applies for a period of three years; which is measured from the date Medicare makes its first pass-through payment for the product. Following the three-year period, the product would be incorporated into the cataract bundled payment system, which could significantly reduce the pricing for that product. Temporary pass-through reimbursement for IHEEZO was awarded by CMS and made effective in the second quarter of 2023 and temporary pass-through reimbursement for TRIESENCE was made effective April 1, 2025. We expect pass-through status for IHEEZO to expire March 31, 2026. Following the expiration of pass-through status, under current CMS policy, non-opioid pain management surgical drugs when used on Medicare Part B patients in an outpatient setting can qualify for ongoing separate payments. CMS’ current non-opioid separate payment policy, like other CMS policies, can be changed by CMS through its annual rulemaking and comment process.
Medicaid is a joint federal and state program that is administered by the states for low-income and disabled beneficiaries. Medicaid rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS, the federal agency that administers the Medicaid and Medicare programs. These data include the average manufacturer price and, in the case of innovator products, the best price for each drug which, in general, represents the lowest price available from the manufacturer to any entity in the U.S. in any pricing structure, calculated to include all sales and associated rebates, discounts, and other price concessions. The amount of the rebate is adjusted upward if the average manufacturer price increases at a faster rate than inflation (measured by reference to the Consumer Price Index – Urban). The rebate was previously capped at 100% of the average manufacturer price, but effective January 1, 2024, this cap on the rebate was removed, and our rebate liability could increase accordingly.
If we become aware that our reporting for a prior quarter was incorrect or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for up to three years after those data originally were due, which revisions could affect our rebate liability for prior quarters. The federal Patient Protection and Affordable Care Act (the “PPACA” or “Health Care Reform Law”) made significant changes to the Medicaid Drug Rebate program, and CMS issued a final regulation, which became effective on April 1, 2016, to implement the changes to the Medicaid Drug Rebate program under the PPACA. Effective in 2022, CMS modified Medicaid Drug Rebate program regulations to, among other things, permit reporting multiple best price figures with regard to value-based purchasing arrangements and provide definitions for “line extension,” “new formulation,” and related terms with the practical effect of expanding the scope of drugs considered to be line extensions.
Civil monetary penalties can be applied if we are found to have knowingly submitted any false pricing or other information to the government, if we are found to have made a misrepresentation in the reporting of our average sales price, or if we fail to submit the required data on a timely basis. Such conduct also could be grounds for CMS to terminate our Medicaid drug rebate agreement, in which case federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs.
Federal law requires that any company that participates in the Medicaid Drug Rebate program also participate in the Public Health Service’s 340B drug pricing program (the “340B program”) in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B program, which is administered by the Health Resources and Services Administration (“HRSA”), requires participating manufacturers to agree to charge statutorily defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. Covered entities include hospitals that serve a disproportionate share of financially needy patients, community health clinics, and other entities that receive certain types of grants under the Public Health Service Act. The PPACA expanded the list of covered entities to include certain free-standing cancer hospitals, critical access hospitals, rural referral centers, and sole community hospitals, but exempts “orphan drugs” from the ceiling price requirements for these covered entities. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and Medicaid rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate program. In general, products subject to Medicaid price reporting and rebate liability are also subject to the 340B ceiling price calculation and discount requirement.
| 11 |
HRSA issued a final regulation regarding the calculation of the 340B ceiling price and the imposition of civil monetary penalties on manufacturers that knowingly and intentionally overcharge covered entities, which became effective on January 1, 2019. It is currently unclear how HRSA will apply its enforcement authority under this regulation. Any charge by HRSA that we have violated the requirements of the regulation could result in civil monetary penalties. Moreover, under a final regulation effective January 13, 2021, HRSA established a new administrative dispute resolution (“ADR”) process for claims by covered entities that a manufacturer has engaged in overcharging, and by manufacturers that a covered entity violated the prohibitions against diversion or duplicate discounts. Such claims are to be resolved through an ADR panel of government officials rendering a decision that could be appealed only in federal court. An ADR proceeding could subject us to onerous procedural requirements and could result in additional liability. On November 30, 2022, HRSA issued a notice of proposed rulemaking that proposes several changes to the ADR process. HRSA also implemented a price reporting system under which we are required to report our 340B ceiling prices to HRSA on a quarterly basis, which then publishes those prices to 340B covered entities. In addition, legislation could be passed that would further expand the 340B program to additional covered entities or would require participating manufacturers to agree to provide 340B discounted pricing on drugs used in an inpatient setting.
In order to be eligible to have our products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we participate in the U.S. Department of Veterans Affairs (“VA”) Federal Supply Schedule (“FSS”) pricing program. FSS participation is required for our products to be purchased by the VA, Department of Defense (“DoD”), Coast Guard, and Public Health Service (“PHS”). Prices for innovator drugs purchased by the VA, DoD, Coast Guard, and PHS are subject to a cap (known as the “Federal Ceiling Price”) equal to 76% of the annual non-federal average manufacturer price (“non-FAMP”) minus, if applicable, an additional discount. The additional discount applies if non-FAMP increases more than inflation (measured by reference to the Consumer Price Index - Urban). We also participate in the Tricare Retail Pharmacy Program, under which we pay quarterly rebates to DoD for prescriptions of our innovator drugs dispensed to Tricare beneficiaries through Tricare Retail network pharmacies. The governing statute provides for civil monetary penalties for failure to provide information timely or for knowingly submitting false information to the government.
Medicare Part D provides coverage to enrolled Medicare patients for self-administered drugs (i.e., drugs that are not administered by a physician). Medicare Part D is administered by private prescription drug plans approved by the U.S. government and, subject to detailed program rules and government oversight, each drug plan establishes its own Medicare Part D formulary for prescription drug coverage and pricing, which the drug plan may modify from time to time. The prescription drug plans negotiate pricing with manufacturers and pharmacies, and may condition formulary placement on the availability of manufacturer discounts. In addition, manufacturers, including us, are required to provide to CMS a 70% discount on brand name prescription drugs utilized by Medicare Part D beneficiaries when those beneficiaries are in the coverage gap phase of the Part D benefit design. The IRA includes a sunset provision with respect to the coverage gap discount program starting in 2025 and replaces it with a new manufacturer discount program. In addition, as of October 2022, the IRA established a Medicare Part D inflation rebate scheme under which, manufacturers will generally owe additional rebates if the average manufacturer price of a Part D drug increases faster than the pace of inflation. Failure to timely pay a Part D inflation rebate is subject to a civil monetary penalty.
Private payor healthcare and insurance providers, health maintenance organizations, and pharmacy benefit managers in the U.S. are adopting more aggressive utilization management techniques and are increasingly requiring significant discounts and rebates from manufacturers as a condition to including products on formulary with favorable coverage and copayment/coinsurance. These payors may not cover or adequately reimburse for use of our products or may do so at levels that disadvantage them relative to competitive products.
| 12 |
Intellectual Property
Our success and ability to compete depends upon our ability to protect our intellectual property. We conduct a fulsome analysis of the intellectual property landscape prior to acquiring rights to formulations and filing patent applications. In addition, as of March 2, 2026, we owned and/or licensed more than 50 issued and pending patent applications, which include U.S.-issued patents, international-issued patents, and U.S. and foreign/international patent pending applications. We expect to file additional patent applications in the U.S. and pursue patent protection for certain of our formulations in other important international jurisdictions in the future.
As of March 2, 2026, we had, on a worldwide basis, more than 100 issued trademarks, pending trademark and copyright applications, or registered copyrights and/or trademarks. We also rely on unpatented trade secrets and know-how and continuing technological innovation in order to develop our products and formulations, which we seek to protect, in part, by confidentiality agreements with our employees, consultants, collaborators and others, including certain service providers. We also have invention or patent assignment agreements with our current employees and certain consultants. However, our employees and consultants may breach these agreements, and we may not have adequate remedies for any breach, or our trade secrets may otherwise become known or be independently discovered by competitors. In addition, inventions relevant to us could be developed by a person not bound by an invention assignment agreement with us, in which case we may have no rights to use the applicable invention.
The following table lists some of our outstanding material patents in the U.S. covering certain branded products we own commercial rights to, general subject matter and latest expiry date. One or more patents with the same or earlier expiry dates may fall under the same general subject matter and are not listed separately.
| Product | General Subject Matter | Expiration | ||
| IHEEZO | Methods using topical formulations Compositions comprising chloroprocaine |
September 2038 May 2039 | ||
| VEVYE | Formulation composition for treatment of dry eye syndrome Ophthalmic composition comprising cyclosporine Semiflourinated compounds for ophthalmic administration Topical administration method |
December 2030 September 2037 November 2038 October 2039 | ||
| TRIESENCE | Composition of injectable suspension Methods for treating ophthalmic disorder |
December 2029 March 2029 | ||
| ILEVRO | Composition comprising carbomer, galactomannan and borate Carboxyvinyle polymer-containing nanoparticle suspension |
December 2030 March 2032 | ||
| BYQLOVI | Aqueous suspension comprising nanoparticles A method of treating an eye inflammatory or infectious disease |
May 2036 May 2036 | ||
| VERKAZIA | Methods for treating eye disease Compositions of oil-in-water cationic emulsion Compositions containing quaternary ammonium compounds |
May 2027 November 2027 June 2029 |
Governmental Regulation
Our business is subject to federal, state and local laws, regulations, and administrative practices, including, among others: federal, state and local licensure and registration requirements concerning the operation of pharmacies and the practice of pharmacy; the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”); the Health Care Reform Law; statutes and regulations of the FDA, the U.S. Federal Trade Commission (the “FTC”), the U.S. Drug Enforcement Administration and the U.S. Consumer Product Safety Commission, as well as regulations promulgated by comparable state agencies concerning the sale, advertisement and promotion of the products we sell. The regulatory and quality compliance environment for compounded drugs has become significantly more rigorous, complex and strict since the passage of The Drug Quality and Security Act of 2013 (the “DQSA”). The complexity of the current state and federal regulatory environment, as well as the expected continued evolution of state and federal laws governing pharmaceutical compounding, have presented, and will continue to present, potentially significant challenges to our business model and the fulfillment of our mission as a company. Below are descriptions of some of the various federal and state laws and regulations which may govern or impact our current and planned operations.
| 13 |
FDA New Drug Application (NDA) Process
As discussed in other sections of this Annual Report, we are pursuing, and may continue to pursue, alone or with project partners, FDA approval to market and sell one or more of our product candidates through the FDA’s NDA process. As a condition of approval, the FDA or other regulatory authorities may require further studies, including Phase 4 post-marketing studies, to provide additional data. Other post-marketing studies may be required to gain approval for the use of a product as a treatment for clinical indications other than those for which the product was initially tested and approved. Also, the FDA or other regulatory authorities require post-marketing reporting to monitor the adverse effects of a drug. Results of post-marketing programs may limit or expand the further marketing of a product.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the Internet. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, fines and potential civil and criminal penalties.
Section 505(b)(2) New Drug Applications
As an alternate path for FDA approval of new indications or new formulations of previously-approved products, a company may file a Section 505(b)(2) NDA instead of a “stand-alone” or “full” NDA. Section 505(b)(2) of the FDCA was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Amendments. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Some examples of products that may be allowed to follow a Section 505(b)(2) path to approval are drugs that have a new dosage form, strength, route of administration, formulation or indication.
The Hatch-Waxman Amendments permit the applicant to rely upon certain published nonclinical or clinical studies conducted for an approved product or the FDA’s conclusions from prior review of such studies. The FDA may require companies to perform additional studies or measurements to support any changes from the approved product. The FDA may then approve the new product for all or some of the labeled indications for which the reference product has been approved, as well as for any new indication supported by the Section 505(b)(2) application. While references to nonclinical and clinical data not generated by the applicant or for which the applicant does not have a right of reference are allowed, all development, process, stability, qualification and validation data related to the manufacturing and quality of the new product must be included in an NDA submitted under Section 505(b)(2).
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s conclusions regarding studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The Section 505(b)(2) application also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its products only to be subject to significant delay and patent litigation before its products may be commercialized.
Pharmacy Regulation
Our pharmacy operations are regulated by both the federal government and the states in which we operate or dispense. State laws and regulations generally address licensing of pharmacists, pharmacy technicians and pharmacies, as well as requirements applicable to compounding activities, including quality standards, sterility assurance, storage, controlled substances, recordkeeping and inspections. State requirements are administered and updated periodically, generally under the jurisdiction of state boards of pharmacy. Failure to comply with applicable state requirements could result in fines, corrective action, heightened oversight, restrictions on operations, suspension, non-renewal or revocation of licenses, or limitations on our ability to dispense into particular jurisdictions. In addition, some states have adopted, or may adopt, more stringent requirements applicable to compounding pharmacies, which could increase compliance costs or limit permissible activities.
| 14 |
The federal regulatory framework for compounding is set forth primarily in Sections 503A and 503B of the FDCA, as amended by the Drug Quality and Security Act (“DQSA”). Section 503A generally addresses pharmacy compounding of patient-specific prescriptions and includes limitations on compounding in advance of receiving prescriptions. Section 503B establishes “outsourcing facilities,” which may compound certain sterile drug products without patient-specific prescriptions, subject to additional requirements, including current good manufacturing practices (“cGMP”) and FDA inspection. Our operations include both a Section 503A pharmacy and a Section 503B outsourcing facility, each subject to the applicable requirements and oversight framework.
Many states require non-resident or out-of-state pharmacies and outsourcing facilities to register with, or obtain licensure from, the applicable state board of pharmacy or other authority to dispense into that state. These requirements vary by state and may change over time. In January 2026, the California Board of Pharmacy approved a settlement agreement between ImprimisRx and the California State Board of Pharmacy, resolving an administrative action brought by the California Board of Pharmacy regarding certain regulatory compliance matters. As part of the settlement, ImprimisRx agreed to surrender its 503B out-of-state outsourcing facility license and its 503A out-of-state compounding pharmacy license on February 1, 2026. If additional states were to take similar actions, or if we are unable to obtain or maintain required registrations or licenses, our ability to dispense compounded products into certain states could be limited and the cumulative effect could be material.
Our Section 503B outsourcing facility is subject to FDA inspection and enforcement authority. We have been subject to FDA inspection and have undertaken remediation and quality initiatives, including engaging an independent third-party cGMP expert. If we are unable to demonstrate sustained compliance with cGMP or other applicable requirements, the FDA could pursue administrative or judicial enforcement actions, which could be costly and could result in restrictions on operations or other adverse consequences. See Item 1A “Risk Factors.”
We compound formulations consistent with applicable standards and requirements, including USP 795 and USP 797, as adopted or applied by regulators, and other applicable state and federal law. Changes in USP standards, FDA policies, state requirements, or enforcement priorities may require operational adjustments and could increase costs or limit permissible activities.
Confidentiality, Privacy and HIPAA
Our pharmacy operations involve the receipt, use and disclosure of confidential medical, pharmacy and other health-related information. In addition, we use aggregated and blinded (anonymous) data for research and analysis purposes. The federal privacy regulations under HIPAA are designed to protect the medical information of a healthcare patient or health plan enrollee that could be used to identify the individual. Among other things, HIPAA limits certain uses and disclosures of protected health information and requires compliance with federal security regulations regarding the storage, utilization and transmission of and access to electronic protected health information. The requirements imposed by HIPAA are extensive. In addition, most states and certain other countries have enacted privacy and security laws that protect identifiable patient information that is not health-related. For example, California enacted the California Consumer Privacy Act (the “CCPA”) that creates new individual privacy rights for consumers and places increased privacy and security obligations on entities handling personal data of consumers or households. Effective January 1, 2020, the CCPA gives California residents expanded privacy rights and protections, and provides civil penalties for violations and a private right of action for data breaches. The CCPA exemplifies the vulnerability of our business to not only cyber threats but also the evolving regulatory environment related to personal data and protected health information. In addition, the California Invasion of Privacy Act prohibits the use of “any machine, instrument, or contrivance” to tap any telephonic communication and use of any “electronic amplifying or recording device” to eavesdrop upon a “confidential communication” without consent of all parties to the communication. Other countries also have, or are developing, laws governing the collection, use and transmission of personal information, such as the General Data Protection Regulation (“GDPR”) in the European Union (the “EU”) that became effective in May 2018 and the Personal Information Protection and Electronic Documents Act that became effective in Canada in April 2000. Further, several states have enacted more protective and comprehensive pharmacy-related privacy legislation that not only applies to patient records but also prohibits the transfer or use for commercial purposes of pharmacy data that identifies prescribers. These regulations impose substantial requirements on covered entities and their business associates regarding the storage, utilization and transmission of and access to personal health and non-health information. Many of these laws apply to our business.
| 15 |
International Regulation
If we pursue commercialization of our products in countries other than the U.S. and where we do not have regulatory market approval, then we may need to obtain the approvals required by the regulatory authorities of such foreign countries that are comparable to the FDA and state boards of pharmacy, and we would be subject to a variety of other foreign statutes and regulations comparable to those relating to our U.S. operations. Regulatory frameworks and requirements vary by country and could involve significant additional licensing requirements and product testing and review periods. We currently partner with companies to sell, market and distribute some of our products in certain foreign countries.
Environmental and Other Matters
We are or may become subject to environmental laws and regulations governing, among other things, any use and disposal by us of hazardous or potentially hazardous substances in connection with our research and preparation of our formulations. In addition, we are subject to work safety and labor laws that govern certain of our operations and our employee relations. In each of these areas, as described above, the FDA and other government agencies have broad regulatory and enforcement powers, including, among other things, the ability to levy fines and civil penalties, suspend or delay issuance of approvals, licenses or permits, seize or recall products, and withdraw approvals, any one or more of which could have a material adverse effect on our business.
Research and Development Expenses
Our R&D expenses incurred in 2025, 2024 and 2023 primarily included expenses related to development of intellectual property, researcher and investigator-initiated evaluations, and formulation development related primarily to our ophthalmic products, formulations and certain other assets, in addition to costs associated with our drug candidate development programs. In 2025, R&D expenses included $8,450,000 recorded as acquired-in-process R&D as part of the acquisition of Melt in November 2025. During the year ended December 31, 2025, we incurred $20,940,000 in R&D expenses, compared to $12,230,000 and $6,652,000 during the year ended December 31, 2024 and 2023, respectively.
Financial Information About Segments and Geographic Areas
The Company has identified two operating segments as reportable segments. The Branded segment includes activities of our FDA approved ophthalmology pharmaceutical products, including the out-licensing of rights to certain of our products. The ImprimisRx segment represents activities in our ophthalmology-focused pharmaceutical compounding business. The Company’s chief operating decision-maker (“CODM”) is the Chief Executive Officer (“CEO”) who evaluates the segment contribution to allocate resources. The CODM does not review segment assets when assessing segment performance and deciding how to allocate resources.
The Company categorizes revenues by geographic area based on selling location. All operations are currently located in the U.S.; therefore, total revenues for 2025, 2024 and 2023 were attributed to the U.S. All long-lived assets at December 31, 2025 and 2024 were located in the U.S.
Human Capital
As of February 25, 2026, we employed 373 individuals. Our employees are engaged in sales, marketing, research, development, pharmacy operations, and general and administrative functions. We expect to add additional employees in all departmental functions, with a focus on sales force additions and other commercial activities as we carry out our business plan in the next 12 months. We are not party to any collective bargaining agreements with any of our employees. We have never experienced a work stoppage, and we believe our employee relations are good. We hire independent contractors and consultants on an as-needed basis.
| 16 |
Talent Acquisition and Retention
We recognize that our employees largely contribute to our success. To this end, we support business growth by seeking to attract and retain best-in-class talent. Our talent acquisition team uses internal and external resources to recruit highly skilled candidates in the U.S. We believe that we continue to attract and retain superior talent as measured by our turnover rate and employee service tenure.
Total Rewards
Our total rewards philosophy has been to create investment in our workforce by offering competitive compensation and benefits packages. We provide employees with compensation packages that include base salary, annual incentive bonuses, and long-term equity awards. We also offer comprehensive employee benefits, which vary by country and region, such as life, disability, and health insurance, health savings and flexible spending accounts, paid time off, and a 401(k) plan. It is our expressed intent to be an employer of choice in our industry by providing market-competitive compensation and benefits packages.
Health, Safety, and Wellness
The health, safety, and wellness of our employees is a priority in which we have always invested and will continue to do so. We provide our employees and their families with access to a variety of innovative, flexible, and convenient health and wellness programs. Program benefits are intended to provide protection and security, so employees can have peace of mind concerning events that may require time away from work or that may impact their financial well-being.
Training and Development
We believe in encouraging employees in becoming lifelong learners by providing ongoing learning, training and leadership opportunities. We provide our employees with a tuition reimbursement program, and in certain instances, onsite training programs. While we strive to provide real-time recognition of employee performance, we have a formal annual review process not only to determine pay and equity adjustments tied to individual contributions, but to identify areas where training and development may be needed.
Company Information
We were incorporated in Delaware in January 2006 as Bywater Resources, Inc. In September 2007, we closed a merger transaction with Transdel Pharmaceuticals Holdings, Inc. and changed our name to Transdel Pharmaceuticals, Inc. As part of a corporate re-organization that was led by our CEO, Mark L. Baum and our President and Chief Financial Officer (“CFO”), Andrew R. Boll, we changed our name to Imprimis Pharmaceuticals, Inc. in February 2012. Then to align with a shift in our corporate strategy that included the expansion into branded ophthalmic products and product candidates, we changed the name of our company to Harrow Health, Inc. in December 2018 and then to Harrow, Inc. in September 2023.
Our corporate headquarters are located at 1A Burton Hills Blvd., Suite 200, Nashville, Tennessee, 37215, and our telephone number at such office is (615) 733-4730.
We file reports with the Securities and Exchange Commission (“SEC”), including Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and other reports from time to time. We are an electronic filer and the SEC maintains an internet site at www.sec.gov that contains the reports, proxy and information statements, and other information filed electronically. Our website address, which is provided as an inactive textual reference only, is www.harrow.com. We make available free of charge through the website Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. Information contained on our website is not deemed part of this Annual Report.
| 17 |
ITEM 1A. RISK FACTORS
Risk Factors Summary
We are subject to a variety of risks and uncertainties, including financial risks, operational risks, human capital risks, legal proceedings and regulatory risks and certain general risks, that could have a material adverse effect on our business results of operations, financial condition and prospects. Risks that we deem material are described below and include, but are not limited to, the following:
Risks Related to Economic Conditions and Operations of Our Business.
| ● | Our ability to achieve and maintain profitability for our business | |
| ● | Our ability to successfully market, commercialize, and sell current, recently acquired and future products | |
| ● | Our current indebtedness and ability to access additional capital | |
| ● | Our ability to attract customers and increase sales of current and future products | |
| ● | Our ability to obtain marketing approval and ongoing expense associated with it for any of our drug candidates, including those for which we own royalty rights | |
| ● | Our reliance on third parties for manufacturing certain components, FDA approved drugs and to conduct clinical trials | |
| ● | Our exposure to liabilities and reputation harm if our products give rise to defects, recalls, patient injury or death | |
| ● | Our information technology systems exposure to cyberattack or information security breach could significantly compromise the confidentiality, integrity and availability of our information technology systems | |
| ● | Global macroeconomic volatility and conditions may adversely affect our business. |
Risks Related to Government Regulations, Trade Policy and Third-Party Policies
| ● | Our business may be affected by litigation, government investigations and injunctive actions | |
| ● | Governmental regulations, including, but not limited to, 503B bulks list and others, that could or currently do burden operations or narrow the market for our products | |
| ● | If we fail to comply with federal or state statutes and regulations with respect to licensure and operation of our business, our pharmacy facilities could be required to cease operations or become subject to restrictions that could adversely affect our business | |
| ● | Our sales depend on coverage and reimbursement from government and commercial third-party payors, and pricing and reimbursement pressures have affected, and are likely to continue to affect, our profitability | |
| ● | The adoption and interpretation of new tax legislation or exposure to additional tax liabilities could affect our profitability | |
| ● | Changes in U.S. trade policy, including tariffs or other import restrictions. |
Risks Related to Competition
| ● | Securing and maintaining patent or other intellectual property protection for our products and related improvements | |
| ● | Market acceptance of our drug products, drug candidates, compounded drugs and pharmacies | |
| ● | Our ability to successfully research, develop and timely manufacture our current and future products and drug candidates | |
| ● | Our ability to enforce protect our intellectual property rights along with the potential of future legal proceedings filed against us claiming intellectual property infringement | |
| ● | Retention, recruitment, and training of senior management and key personnel |
| 18 |
Risks Related to Product Development, Regulatory Approval, Manufacturing and Commercialization
| ● | We may not be able to develop commercial products despite significant investments in R&D | |
| ● | Our branded products and product candidates in development cannot be sold without regulatory approval | |
| ● | Our drug candidates may face competition sooner than we expect | |
| ● | We rely on third parties to manufacture and conduct clinical trials of our branded drug products and product candidates | |
| ● | We may not be successful in obtaining market exclusivity for our product candidates | |
| ● | Disruptions at government agencies—due to funding lapses, shutdowns, staffing constraints, reorganizations, or shifting policy priorities—could adversely affect our business . |
Risks Related to Our Indebtedness
| ● | Our ability to pay interest and debt service payments associated with the 2030 Notes (as defined below) | |
| ● | We could enter into various transactions that could increase the amount of our outstanding debt or adversely affect our capital structure or credit rating. |
Risks Related to Our Common Stock
| ● | Volatility of the price of our common stock | |
| ● | Our stock price falling as a result of future offerings or sales |
You should carefully consider the following risk factors in addition to the other information contained in this Annual Report. Our business, financial condition, results of operations, and prices of our common stock could be materially adversely affected by any of these risks.
Risks Related to Economic Conditions and Operations of Our Business.
We may not be profitable in the future.
As of December 31, 2025, our accumulated deficit was $156,524,000. Our current projections indicate that we will have operating income and/or net income during 2026; however, these projections may not be correct and our plans could change. Also, we could incur operating losses in the foreseeable future for our commercialization activities, research and development, and our pharmaceutical compounding business, which would impact net income. Although we have been generating revenue from our operations, our ability to generate the revenues necessary to achieve and maintain profitability will depend on many factors, including those discussed in this “Risk Factors” section. Our business plan and strategies involve costly activities that are susceptible to failure, and, therefore, we may not be able to generate sufficient revenue to support and sustain our business or reach the level of sales and revenues necessary to achieve and sustain profitability.
We may not receive sufficient revenue to fund our operations and recover our development costs.
Our business plan involves the sale and marketing of FDA-approved products, compounded formulations and development of drug candidates through third-party wholesaler and pharmacy channels and our ImprimisRx facilities. We have limited experience selling FDA-approved products, and we may be unable to successfully manage this business or generate sufficient revenue to recover our development costs and operational expenses. We may have only limited success in marketing and selling our products. Although we have established and plan to grow our internal sales teams to market and sell our products, we have limited experience with such activities and may not be able to generate sufficient physician and patient interest in our products to generate significant revenue from sales of these products.
We may fail to realize the anticipated benefits of our recent and any future product acquisitions.
The success of our product acquisitions will depend on, among other things, our ability to integrate the products into our commercial platform, transfer the products NDAs, maintain and obtain sufficient payor reimbursement coverage, maintain an adequate supply of the products, market the products to our existing customers and re-introduce BYOOVIZ to the ophthalmic market. If we experience difficulties with the implementation of plans with respect to our acquisitions, the anticipated benefits of recent or future acquisitions may not be realized fully or at all, or may take longer to realize than expected. Integration efforts will also divert management’s attention and resources. These matters could have an adverse effect during any transition period and for an undetermined period after completion of the acquisitions.
| 19 |
We may not be able to correctly estimate our future operating expenses, which could lead to cash shortfalls.
The estimates of our future operating and capital expenditures are based upon our current business plan, our current operations and our current expectations regarding product sales. Our projections have varied significantly from actual performance in the past as a result of changes to our business model, strategy and acquisitions. We may not accurately estimate the potential revenues and expenses of our operations. If we are unable to correctly estimate the amount of cash necessary to fund our business, we could spend our available financial resources much faster than we expect. If we do not have sufficient funds to continue to operate and develop our business, we could be required to seek additional financing earlier than we expect, which may not be available when needed or at all, or be forced to delay, scale back or eliminate some or all of our proposed operations.
If we do not successfully identify and acquire rights to new products and drug candidates and successfully integrate them into our operations, our growth opportunities may be limited.
We plan to pursue the development of new FDA approved products and drug candidates which may include continued activities to develop and commercialize current assets or, if and as opportunities arise, potential acquisitions of new intellectual property rights and assets. We have historically relied, and, to a certain extent, we expect to continue to rely, primarily upon third parties to provide us with additional development opportunities. We may seek to enter into acquisition agreements or licensing arrangements to obtain rights to develop new formulations and FDA approved products in the future, but only if we are able to identify attractive products and formulations and negotiate acquisition or license agreements on terms acceptable to us, which we may not be able to do. Moreover, we have limited resources to acquire additional potential product development assets and integrate them into our business. Acquisition opportunities may involve competition among several potential purchasers, which could include large multi-national pharmaceutical companies and other competitors that have access to greater financial resources than we do. If we are unable to obtain rights to development and commercial opportunities from third parties and we are unable to rely upon our compounding pharmacies and current and future relationships with pharmacists, physicians and other inventors to provide us with additional development opportunities, our growth and prospects could be limited.
Our product development strategy is to focus on ophthalmology and eye care related products for which we believe there is market potential, unmet needs and/or unique value to physicians and patients and to develop and offer products within these therapeutic areas that could afford us with gross and operating margins consistent with our current and historical figures. However, our expectations and assumptions about market potential and patient needs may prove to be wrong, and we may invest capital and other resources on products, drug candidates, and formulations that do not generate sufficient revenues for us to recoup our investment.
We may be unable to successfully develop and commercialize our drug products, candidates or any other assets we may acquire.
We have acquired assets related to drug products and drug candidates. We are currently pursuing development and commercialization opportunities with respect to a number of these products and drug candidates, and we are in the process of assessing certain of our other assets in order to determine whether to pursue their development or commercialization. In addition, we expect to consider the acquisition of additional intellectual property rights or other assets in the future. Once we decide to pursue a potential drug candidate, we develop a commercialization strategy for it, which may include pursuing FDA approval of the drug candidate. We may incorrectly assess the risks and benefits of the commercialization options or we may not pursue a commercialization strategy that proves to be successful. If we are unable to successfully commercialize one or more of our drug products and drug candidates, our operating results would be adversely affected. Even if we are able to successfully sell one or more drug products and drug candidates, we may never recoup our investment in acquiring or developing the drug products and drug candidates. Our failure to identify and expend our resources and technologies with commercial potential and execute an effective commercialization strategy for each of our drug products and drug candidates would negatively impact the long-term profitability of our business.
| 20 |
We may need additional capital in order to continue operating our business and to operate as a going concern, and such additional funds may not be available when needed, on acceptable terms, or at all.
We may need significant additional capital to execute our business plan, execute on future acquisitions and fund our proposed business operations. Additionally, our plans may change or the estimates of our operating expenses and working capital requirements could be inaccurate, we may pursue acquisitions of FDA-approved products, drug candidates, pharmacies or other strategic transactions that involve large expenditures, or we may experience growth more quickly or on a larger scale than we expect, any of which may result in the depletion of capital resources more rapidly than anticipated and could require us to seek additional financing earlier than we expect to support our operations.
We may seek to obtain additional capital through equity or debt financings, funding from corporate partnerships or licensing arrangements, sales of assets or other financing transactions. If we issue additional equity or convertible debt securities to raise funds, our existing stockholders may experience substantial dilution, and the newly issued equity or debt securities may have more favorable terms or rights, preferences and privileges senior to those of our existing stockholders. If we raise additional funds through collaboration and licensing arrangements or sales of assets, we may have to relinquish potentially valuable rights to our drug candidates or proprietary technologies, or grant licenses on terms that are not favorable to us. If we raise funds by incurring additional debt, we may be required to pay significant interest expenses and our leverage relative to our earnings or to our equity capitalization may increase. Obtaining commercial loans, assuming those loans would be available, would increase our liabilities and future cash commitments and may impose restrictions on our activities, such as the financial and operating covenants. Further, we may incur substantial costs in pursuing future capital and/or financing transactions, including investment banking fees, legal fees, accounting fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we may issue, such as options, convertible notes and warrants, which would adversely impact our financial results.
We have in the past participated and may in the future participate in strategic transactions that could impact our liquidity, increase our expenses and distract our management.
From time to time, we consider engaging in strategic transactions, such as out-licensing or in-licensing of compounds, drug candidates, drug products or technologies, acquisitions of companies, and asset purchases. We may also consider a variety of different business arrangements in the future, including strategic partnerships, joint ventures, spin-offs, carve-outs, restructurings, divestitures, business combinations and investments. In addition, another entity may pursue us or certain of our assets or aspects of our operations as an acquisition target. Any such transactions may require us to incur expenses specific to the transaction and not incident to our operations, may increase our near- and long-term expenditures, may pose significant integration challenges, may require us to hire or otherwise engage personnel with additional expertise, or may result in our selling or licensing of our assets or technologies under terms that may not prove profitable, any of which could harm our operations and financial results. Such transactions may also entail numerous other operational and financial risks, including, among others, exposure to unknown liabilities, disruption of our business and diversion of our management’s time and attention in order to develop acquired products, drug candidates, technologies or businesses.
As part of our efforts to complete any significant transaction, we would need to expend significant resources to conduct business, regulatory, legal and financial due diligence, with the goal of identifying and evaluating material risks involved in the transaction. We may be unsuccessful in ascertaining or evaluating all the risks and, as a result, we may not realize the expected benefits of the transaction, whether due to unidentified risks, integration difficulties, regulatory setbacks or other events. We may incur material liabilities for the past activities of any businesses we partner with or acquire. If any of these events occur, we could be subject to significant costs and damage to our reputation, business, results of operations and financial condition.
| 21 |
If we are unable to establish, train and maintain an effective sales and marketing infrastructure, we will not be able to commercialize our drug products and candidates successfully.
We have built an internal sales and marketing infrastructure to implement our business plan by developing internal sales teams and education campaigns to market our drug products. We will need to expend significant resources to further establish and grow this internal infrastructure and properly train sales personnel with respect to regulatory compliance matters. We may also choose to engage or enter into other arrangements with third parties to provide sales and marketing services for us in place of or to supplement our internal commercialization infrastructure. We may not be able to secure sales personnel or relationships with third-party sales organizations that are adequate in number or expertise to successfully market and sell our proprietary formulations, drug products and pharmacy services. Further, any third-party organizations we may seek to partner with or engage may not be able to provide sales and marketing services in accordance with our expectations and standards, may be more expensive than we can afford or may not be available on otherwise acceptable terms or at all. If we are unable to establish and maintain compliant and adequate sales and marketing capabilities, through our own internal infrastructure or third-party services or other arrangements, we may be unable to sell our formulations, drug products or services or generate meaningful revenues.
We depend upon consultants, outside contractors and other third-party service providers for key aspects of our business.
We are substantially dependent on consultants and other outside contractors and service providers for key aspects of our business. For instance, we rely upon pharmacist, physician and research consultants and advisors to provide us with significant assistance in the evaluation of product development opportunities, and we have engaged or supported, and expect to continue to engage or support, consultants, advisors, contract manufacturers, clinical research organizations (“CROs”), and others to design, conduct, analyze and interpret the results of any clinical or non-clinical trials or other studies in connection with the research and development of our products. If any of our consultants or other service providers terminates its engagement with us, or if we are unable to engage highly qualified replacements as needed on commercially reasonable terms, we may be unable to successfully execute our business plan. We must effectively manage these third-party service providers to ensure that they successfully carry out their contractual obligations and meet expected deadlines. However, these third parties often engage in other business activities and may not devote sufficient time and attention to our activities, and we may have only limited contractual rights in connection with the conduct of the activities we have engaged the service providers to perform. If we are unable to effectively manage our outsourced activities or if the quality, timeliness or accuracy of the services provided by third-party service providers is compromised for any reason, our development activities may be extended, delayed or terminated, and we may not be able to commercialize our formulations or advance our business.
If any of our products, compounded formulations, or product candidates cause, or are alleged to have caused, patient injury or death, or are subject to a recall or other corrective action, we could face significant liabilities and reputational harm
Our business depends on the safety, quality, and performance of our products and services, as well as the perceptions of physicians, patients, regulators, and other stakeholders. We market and sell FDA-approved branded pharmaceutical products, we provide compounded formulations through our pharmacy operations, and we are developing additional product candidates. Any actual or perceived safety, quality, labeling, manufacturing, storage, distribution, or use-related issue involving any of our products, compounded formulations, or product candidates—including contamination, sterility failures, defects, adverse events, medication errors, misuse, off-label use, inadequate warnings, or quality concerns related to raw materials or components—could result in negative publicity, product complaints, regulatory scrutiny, loss of customer confidence, reduced demand, and harm to our reputation.
In addition, we may be subject to voluntary or involuntary recalls, market withdrawals, field corrections, “Dear Healthcare Provider” letters, product discontinuations, or other corrective actions, whether initiated by us, required by the FDA, state boards of pharmacy, or other regulators, or prompted by third parties in our supply chain. Such events could also lead to inspections, warning letters, enforcement actions, increased oversight, remediation costs, and, with respect to our pharmacy operations, the suspension, restriction, non-renewal, or loss of pharmacy licenses or registrations in one or more jurisdictions. Even if a safety or quality event ultimately is determined to be unrelated to our products or results from a false positive, isolated incident, or a third party’s actions, the resulting reputational and operational impacts could be significant.
If any of these events occur, we could incur substantial costs, including product and professional liability claims, litigation and settlement expenses, regulatory penalties, remediation and recall costs, increased insurance costs (or reduced availability of insurance), and lost revenue due to reduced prescribing, reduced utilization, supply disruption, or delays in development and commercialization. Any such outcomes could materially and adversely affect our business, financial condition, results of operations, and cash flows.
| 22 |
We carry product and professional liability insurance, which may be inadequate.
Although we have secured product and professional liability insurance for our products, pharmacy operations and the marketing and sale of our formulations, our current or future insurance coverage may prove insufficient to cover any liability claims brought against us. Because of the increasing costs of insurance coverage, we may not be able to maintain insurance coverage at a reasonable cost or at a level adequate to satisfy liabilities that may arise.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of CROs, contractors and consultants, could be subject to power shortages, telecommunications failures, wildfires, water shortages, floods, earthquakes, hurricanes, typhoons, fires, extreme weather conditions, public health crises, and other natural or man-made disasters or business interruptions for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of our contract manufacturers or the contract manufacturers of our development partners are affected by a man-made or natural disaster or other business interruption.
Our compounding operations and our broader pharmaceutical business are subject to significant regulatory, quality, and commercial risks, and we may not achieve our objectives for these operations.
Our operations are subject to extensive federal and state regulation and oversight. This includes licensure and operational requirements applicable to our pharmacy and compounding activities, and FDA and other regulatory requirements applicable to the development, manufacture, labeling, promotion, and distribution of our FDA-approved branded products and product candidates. Failure to obtain, maintain, renew, or comply with required licenses, registrations, approvals, permits, or quality standards, or changes in applicable laws, regulations, guidance, or enforcement priorities, could restrict or delay our ability to operate in one or more jurisdictions, delay development or commercialization activities, increase compliance and remediation costs, or result in warnings, inspections, product seizures, recalls, injunctions, civil or criminal penalties, or other enforcement actions.
Compounded formulations are not FDA-approved drug products and may face limited acceptance, reimbursement constraints, and competition from FDA-approved alternatives. Negative publicity or regulatory activity involving compounding pharmacies (whether or not involving us) could also reduce demand for compounded products. We have previously received FDA regulatory observations and communications relating to our compounding operations, including with respect to our 503B outsourcing facility and certain pharmacy operations, and we continue to invest in compliance and quality initiatives. See “We have been in discussions with the FDA regarding past inspections of our 503B facility, and to the extent we are unable to demonstrate compliance with cGMPs and other required regulations, the government could pursue enforcement actions, the effects of which could be costly to us and could result in adverse consequences to our business.”
Our FDA-approved branded products and product candidates are subject to significant regulatory and commercial risks, including risks related to manufacturing quality and supply chain reliability, labeling and promotional restrictions, competitive dynamics (including from generics, biosimilars, and alternative therapies), and pricing and reimbursement pressures. We may incur significant costs to maintain and enhance quality systems, satisfy regulatory requirements, and respond to inspections or other regulatory inquiries, and these efforts may divert resources from other parts of our business and may not produce the intended benefits. If physicians, patients, or third-party payors are unwilling to prescribe, use, or reimburse our compounded formulations, branded products, or future product candidates, or if our operations are constrained by regulatory actions, licensure limitations, or increased compliance burdens, our business, financial condition, results of operations, and cash flows could be materially and adversely affected.
| 23 |
A breakdown of our information technology systems, or a cyberattack or information security breach could significantly compromise the confidentiality, integrity and availability of our information technology systems, network-connected control systems and/or our data, interrupt the operation of our business, affect our reputation and result in remediation costs, litigation, or regulatory inquiries.
To achieve our business objectives, we rely on sophisticated information technology systems, including hardware, software, technology infrastructure, online sites and networks for both internal and external operations, mobile applications, cloud services and network-connected control systems, some of which are managed, hosted, provided or serviced by third parties. Internal or external events that compromise the confidentiality, integrity and availability of our systems and data may significantly interrupt the operation of our business, result in significant costs and/or adversely affect our reputation.
Our information technology systems are highly integrated into our business, including our customer service infrastructure, R&D efforts, clinical and commercial manufacturing processes and product sales and distribution processes. Further, as the large part of our employees work remotely for some portion of their jobs, our reliance on our third-party information technology systems has increased substantially and is expected to continue to increase. Remote and hybrid working arrangements can increase cybersecurity risks due to the challenges associated with managing remote computing assets and security vulnerabilities that are present in many non-corporate and home networks. The complexity and interconnected nature of software, hardware and our systems make them vulnerable to breakdown or other service interruptions, and to software errors or defects, misconfiguration and other security vulnerabilities. Upgrades or changes to our systems or the software that we use have resulted and we expect, in the future, will result in the introduction of new cybersecurity vulnerabilities and risks. Our systems are also subject to frequent perimeter network reconnaissance and scanning, phishing and other cyberattacks. As the cyber-threat landscape evolves, these attacks are growing in frequency, sophistication, and intensity, and are becoming increasingly difficult to detect and increasingly sophisticated in using techniques and tools—including artificial intelligence—that circumvent security controls, evade detection and remove forensic evidence. Such attacks could include the use of harmful and virulent malware, including ransomware or other denials of service, which can be deployed through various means, including the software supply chain, e-mail, malicious websites and/or the use of social engineering/phishing.
We have experienced attacks against our network, although none that have had a material adverse impact to our business. In November 2024, we became aware of a cybersecurity incident that involved unauthorized access of an employee’s email account. Through this unauthorized access the threat actor was able to fraudulently divert Company funds to its bank account. We detected the incident in a timeframe management believes minimized the financial, operation or reputational risk to the Company, and at no point was our ability to generate revenues disrupted. However, if future attacks occur, there is no assurance we will be able to detect the incident in a timely manner or at all.
There can be no assurance that our efforts to guard against the wide and growing variety of potential attack techniques will be successful. Attacks such as those experienced by government entities (including those that approve and/or regulate our products) and other multi-national companies, including some of our peers, could leave us unable to utilize key business systems or access or protect important data, and could have a material adverse effect on our ability to operate our business, including developing, gaining regulatory approval for, manufacturing, selling and/or distributing our products. For example, in 2017, a pharmaceutical company experienced a cyberattack involving virulent malware that significantly disrupted its operations, including its research and sales operations and the production of some of its medicines and vaccines. As a result of the cyberattack, its orders and sales for certain products were negatively affected. In late 2020, SolarWinds Corporation, a leading provider of software for monitoring and managing information technology infrastructure, disclosed that it had suffered a cybersecurity incident whereby attackers had inserted malicious code into legitimate software updates for its products that were installed by myriad private and government customers, enabling the attackers to access a backdoor to such systems. In 2022, Okta, Inc., a provider of software that helps companies manage user authentication, disclosed that several hundred of its corporate customers were vulnerable to a security breach that allowed attackers to access Okta’s internal network. Although this breach did not have a significant effect on our business, there can be no assurance that a similar future breach would not result in a material adverse effect on our business or results of operations.
| 24 |
Our systems contain and use a high volume of sensitive data, including intellectual property, trade secrets and other proprietary business information, financial information, regulatory information, strategic plans, sales trends and forecasts, litigation materials and/or personal identifiable information belonging to us, our staff, our patients, customers and/or other parties. In some cases, we utilize third-party service providers to collect, process, store, manage or transmit such data, which have increased our risk. Intentional or inadvertent data privacy or security breaches (including cyberattacks) resulting from attacks or lapses by employees, service providers (including providers of information technology-specific services), business partners, nation states (including groups associated with or supported by foreign intelligence agencies), organized crime organizations, “hacktivists” or others, create risks that our sensitive data may be exposed to unauthorized persons, our competitors or the public. System vulnerabilities and/or cybersecurity breaches experienced by our third-party service providers constitute a substantial share of the information security risks to our business. There can be no assurance that a cybersecurity incident would not result in a material adverse effect on our business or results of operations. Further, the timeliness of our awareness of a cybersecurity incident affects our ability to respond to and work to mitigate the severity of such events.
Cyberattackers are also increasingly exploiting vulnerabilities in commercially available software from shared or open-source code. We rely on third party commercial software that have had and may have such vulnerabilities, but as use of open-source code is frequently not disclosed, our ability to fully assess this risk to our systems is limited. There can be no assurances that a vulnerability in the software and services that we use would not result in a material adverse effect on our business or results of operations.
Domestic and global government regulators, our business partners, suppliers with whom we do business, companies that provide us or our partners with business services and companies we have acquired or may acquire face similar risks. Security breaches of their systems or service outages have adversely affected systems and could, in the future, affect our systems and security, leave us without access to important systems, products, raw materials, components, services or information, or expose our confidential data or sensitive personal information. An extended service outage affecting these or other vendors, particularly where such vendor is the single source from which we obtain the services, could have a material adverse effect on our business or results of operations. For example, in February 2024, UnitedHealth Group announced that a suspected nation-state associated cybersecurity threat actor had gained access to some of the Change Healthcare (“Change”) information technology systems. Change is the largest clearinghouse for medical claims in the U.S. While Harrow was not directly impacted by this cybersecurity incident, it was reported that as a reaction to the cybersecurity incident, Change temporarily disconnected over 100 related payment systems and Change was unable to process medical claims through its primary platforms. This resulted in the delays to the revenue and cash collection cycle for several ASCs and physician offices, putting a strain on their cash resources. While temporary, the cash constraints for these ASCs and physician offices, we believe, impacted sales of some of our products, such as IHEEZO, during this disrupted period of time. In addition, we distribute our products in the U.S. primarily through three pharmaceutical wholesalers, and a security breach that impairs the distribution operations of our wholesalers could significantly impair our ability to deliver our products to healthcare providers and patients. There can be no assurance that our cybersecurity risk management program and processes, including our policies, controls, or procedures, will be effective in protecting our information technology systems and sensitive data.
We will continue to experience varying degrees of cyberattacks and other incidents in the future. Even though we continue to invest in the monitoring, protection and resilience of our critical and/or sensitive data and systems, there can be no assurances that our efforts will detect, prevent or fully recover systems or data from all breakdowns, service interruptions, attacks and/or breaches of our systems that could adversely affect our business and operations and/or result in the loss or exposure of critical, proprietary, private, confidential or otherwise sensitive data, which could result in material financial, legal business or reputational harm to us or negatively affect our stock price. While we maintain cyber-liability insurance, our insurance is not sufficient to cover us against all losses that could potentially result from a service interruption, breach of our systems or loss of our critical or sensitive data.
We are also subject to various laws and regulations globally regarding privacy and data protection, including laws and regulations relating to the collection, storage, handling, use, disclosure, transfer and security of personal data. The legislative and regulatory environment regarding privacy and data protection is continuously evolving and developing and the subject of significant attention globally. For example, we are subject to the CCPA, which became effective in January 2020, which can result in substantial penalties for noncompliance. The CCPA was amended in late 2020, to create the California Privacy Rights Act to create opt-in requirements for the use of sensitive personal data and the formation of a new dedicated agency for the enforcement of the law, the California Privacy Protection Agency. Similar consumer privacy laws went into effect in Virginia, Colorado, Utah, Connecticut and Florida in 2023. Consumer privacy laws were also passed in 11 other states, with the earliest effective dates later this year, and proposed in three additional states. Failure to comply with these current and future laws could result in significant penalties and reputational harm and could have a material adverse effect on our business and results of operations.
| 25 |
Our current and future use of artificial intelligence (“AI”), including by third-party vendors we rely on, may create new operational, regulatory, legal, and reputational risks, and may adversely affect our business.
We and certain third parties that support our business may use artificial intelligence and machine learning technologies, including generative AI tools, to automate or enhance workflows, analyze data, and support decision-making across functions. These technologies are rapidly evolving and can be difficult to evaluate and control, and they may produce inaccurate, incomplete, misleading, or biased outputs that are not readily detectable. If AI-supported processes, analyses, or content are flawed or used inappropriately, we could experience operational inefficiencies, delays, or other adverse impacts.
Because we operate in highly regulated markets, the use of AI may heighten our compliance and liability risks. For example, AI-assisted materials used in commercial, medical, quality, complaint handling, or safety-related activities could contain errors or omit context, and inadequate governance, documentation, or human oversight could increase regulatory scrutiny, remediation costs, or reputational harm. In addition, reliance on third parties to develop, implement, or operate AI-enabled systems may reduce our visibility into how outputs are generated and how controls are applied.
AI tools can also increase confidentiality, cybersecurity, privacy, and intellectual property risks. Use of third-party AI platforms by our employees or service providers may result in unintended disclosure, processing, or use of confidential information, proprietary know-how, or personal data, and may increase exposure to cyber threats, including social engineering. Further, laws, regulations, and enforcement priorities relating to AI are developing rapidly and may be inconsistent across jurisdictions, which could increase compliance costs, restrict certain uses, or require changes to our controls; conversely, if we do not adopt AI effectively relative to competitors, we may be disadvantaged in efficiency and speed.
Global macroeconomic conditions could adversely affect our business and results of operations.
Macroeconomic volatility—including changes in inflation, interest rates, credit availability, and broader financial market conditions—may adversely affect our business. These conditions can reduce demand for healthcare services, increase the portion of costs borne by patients, and pressure third-party payors’ budgets, which may negatively affect utilization of and reimbursement for our products.
Macroeconomic and geopolitical developments may also increase or destabilize our operating costs, including labor, freight, energy, and materials, and may contribute to supply chain disruption, vendor constraints, or counterparty financial distress. In addition, changes in trade policy and international conflicts may create further uncertainty and cost pressure. If these conditions persist or worsen, our sales, operating expenses, and results of operations could be adversely affected.
Risks Related to Government Regulations and Third-Party Policies
Our business is significantly impacted by state and federal statutes and regulations.
The marketing and sale of our drug candidates, FDA-approved drugs and compounded formulations are subject to and must comply with extensive state and federal statutes and regulations governing those products and compounding pharmacies. These compounding statutes and regulations include, among other things, restrictions on compounding for office use or in advance of receiving a patient-specific prescription or, for outsourcing facilities, requirements regarding preparation, such as regular FDA inspections and cGMP requirements, prohibitions on compounding drugs that are essentially copies of FDA-approved drugs, limitations on the volume of compounded formulations that may be sold across state lines, and prohibitions on wholesaling or reselling. These and other restrictions on the activities of compounding pharmacies and outsourcing facilities may significantly limit the market available for compounded formulations, compared to the market available for FDA-approved drugs.
| 26 |
Our pharmacy business is impacted by federal and state laws and regulations governing the following: the purchase, distribution, management, compounding, dispensing, reimbursement, marketing and labeling of prescription drugs and related services including: FDA and/or state regulation affecting the pharmacy and pharmaceutical industries, including state pharmacy licensure and registration or permit standards; rules and regulations issued pursuant to HIPAA and other state and federal laws related to the use, disclosure and transmission of health information; and state and federal controlled substance laws. Our failure to comply with any of these laws and regulations could severely limit or curtail our pharmacy operations, which would materially harm our business and prospects. Further, our business could be adversely affected by changes in these or any newly enacted laws and regulations, and federal and state agency interpretations of the statutes and regulations. Statutory or regulatory changes could require us to make changes to our business model and operations and/or could require us to incur significantly increased costs to comply with such regulations.
On July 30, 2020, the FDA issued a notice for comments related to certain bulk drug substances to be removed from the 503B Bulk’s List (or Category 1 List). Included in this notice for comment were certain bulk drug substances which we currently use in some of our compounded products. In the event one or more of these bulk substances are ultimately removed from the Category 1 List, we intend to utilize commercially available versions of these substances or similar active pharmaceutical ingredients (“APIs”) as replacements of the bulk powders contained in our sterile products. In addition, nothing in the FDA’s notice affects the dispensing of bulk powder-containing products from our 503A pharmacy. Nonetheless, if all or some of the bulk drug substances we use are removed from the 503B Bulk’s List, this may result in a disruption in our operations, revenues and cash flows.
Federal and state compounding frameworks—including evolving FDA policies regarding interstate distribution and information-sharing—may change and could impose additional compliance obligations or restrict aspects of our compounding operations, which could adversely affect our results of operations.
We have been in discussions with the FDA regarding past inspections of our 503B facility, and to the extent we are unable to demonstrate compliance with cGMPs and other required regulations, the government could pursue enforcement actions, the effects of which could be costly to us and could result in adverse consequences to our business.
We have received inspection observations, warning letters and other regulatory communications in the past from the FDA, and we continue to invest in quality systems, remediation activities, and compliance resources, including third-party support. If we are unable to maintain compliance, the FDA or other regulators could pursue enforcement actions, which may include additional warning letters, product seizures, recalls, restrictions on manufacturing or distribution, consent decrees, injunctions, civil or criminal penalties, or other measures. Any such actions could increase costs, disrupt operations, and negatively affect our reputation, results of operations, and financial condition.
From March 2024 through April 2024, NJOF was inspected by the FDA (the “2024 Inspection”), and the FDA issued a Form 483 with five observations. During the first quarter of 2025, we engaged in separate but related discussions with the federal government regarding the NJOF quality system and the 2024 Inspection. NJOF voluntarily recalled certain products and provided regular updates to the FDA regarding its remediation activities and other commitments, including Project Beagle (as discussed in Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations – Recent Developments.”). The government has notified us that these discussions are now closed. In October 2025, ImprimisRx, had an in-person meeting with the FDA to further present and discuss its efforts to remediate certain deficiencies at NJOF. At this meeting the FDA ultimately decided to allow ImprimisRx to continue with its remediation efforts on a voluntary basis with ImprimisRx providing a commitment to ongoing communication regarding its remediation efforts with the FDA.
Future regulatory actions could increase scrutiny and could create negative publicity on us as a company. As part of our commitment to actively work with regulators, at times, we have become aware of concerns related to certain formulations, and as a result, discontinued compounding certain drug formulations in an attempt to help mitigate potential regulatory risk. Physicians may be unwilling to prescribe or patients may be unwilling to use our compounded formulations for other reasons, including but not limited to, the following: legal prohibitions on our ability to discuss the efficacy or safety of our formulations with potential users to the extent applicable data is available; our pharmacy operations are primarily operating on a cash-pay basis and reimbursement may or may not be available from third-party payors, including the government Medicare and Medicaid programs; and certain formulations are not required to be prepared and are not presently being prepared in a manufacturing facility governed by cGMP requirements. These factors and any future regulatory action could continue to limit our production, and our ability to dispense and distribute our compounded products, which would negatively affect sales of our compounded products.
| 27 |
If we (or a partner facility) fail to comply with the Controlled Substances Act, FDCA, or similar state statutes and regulations, the pharmacy facilities could be required to cease operations or become subject to restrictions that could adversely affect our business.
Our pharmacy operations are subject to extensive federal and state laws and regulations. Many states require pharmacies that dispense pharmaceuticals into the state to be appropriately licensed or registered, and state controlled substance laws generally require registration and compliance with standards promulgated by state pharmacy and controlled substance authorities. These laws and regulations often address the qualifications of personnel, prescription fulfillment and inventory control practices, facility standards, recordkeeping, and related compliance obligations, and they subject pharmacies to oversight by state boards of pharmacy and other regulators that may impose corrective actions, enhanced oversight, or operational restrictions if a pharmacy is found not in compliance.
While we maintain compliance and quality programs intended to support adherence to applicable requirements, if any of our pharmacy operations (or a partner facility on which we rely) fail to comply with applicable laws or regulations, the affected operations could be required to temporarily or permanently limit or cease dispensing or compounding activities, which could adversely affect our ability to provide compounded formulations and could materially harm our business. Noncompliance could also result in adverse actions by state boards of pharmacy or other regulators. In addition, FDA inspections and related enforcement under the FDCA, including with respect to the exemptions applicable to compounders under Sections 503A and 503B, could result in warning letters, injunctions, fines, loss of exemptions, or other enforcement actions, any of which could involve significant costs and could adversely affect our business.
ImprimisRx was subject to an administrative action by the California State Board of Pharmacy relating to certain regulatory compliance matters and entered into a settlement to resolve that matter in November, 2025. As part of the settlement, effective February 1, 2026, ImprimisRx voluntarily surrendered its California nonresident 503B outsourcing facility license and California nonresident 503A compounding pharmacy license. The loss of these licenses restricts our ability to sell or dispense certain compounded products in California and has reduced revenues from that state. We believe that, based on our current sales concentration and geographic diversification, this loss is not material to our consolidated statement of operations. However, if additional states were to take similar actions, or if broader or longer-term restrictions were imposed, the cumulative effect could be material.
If we market any of our drug candidates in a manner that violates healthcare fraud and abuse laws, or if we violate government price reporting laws, we may be subject to civil or criminal penalties.
The FDA enforces laws and regulations which require that the promotion of pharmaceutical products be consistent with the approved prescribing information. While physicians may prescribe an approved product for a so-called “off label” use, it is unlawful for a pharmaceutical company to promote its products in a manner that is inconsistent with its approved label, and any company which engages in such conduct can subject that company to significant liability. Similarly, industry codes in the EU and other foreign jurisdictions prohibit companies from engaging in off-label promotion, and regulatory agencies in various countries enforce violations of the code with civil penalties. While we intend to ensure that our promotional materials are consistent with our label, regulatory agencies may disagree with our assessment and may issue untitled letters, warning letters or may institute other civil or criminal enforcement proceedings. In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal healthcare fraud and abuse laws have been applied in recent years to restrict certain marketing practices in the pharmaceutical industry. These laws include the U.S. Anti-Kickback Statute, U.S. False Claims Act and similar state laws. Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of these laws.
| 28 |
Our sales depend on coverage and reimbursement from government and commercial third-party payors, and pricing and reimbursement pressures have affected, and are likely to continue to affect, our profitability.
Sales of our products depend on the availability and extent of coverage and reimbursement from third-party payors, including government healthcare programs and private insurance plans. Payors continue to implement measures to manage utilization and contain costs, including step edits, prior authorization, formulary restrictions, increased patient cost sharing, and reimbursement rate reductions. These actions may reduce the number of patients for whom our products are reimbursed, delay or restrict patient access, and limit our ability to increase prices or maintain pricing levels, any of which could adversely affect our revenues and profitability.
In the U.S., legislative and regulatory actions continue to focus on reducing drug costs, including measures affecting Medicare reimbursement and manufacturer financial obligations. In addition, policymakers and CMS have advanced proposals that would reference prices in other economically comparable countries in determining Medicare beneficiary cost-sharing and/or additional manufacturer rebate obligations (sometimes described as “most-favored-nation” or international reference pricing policies). CMS has proposed mandatory demonstration models—GLOBE (Medicare Part B) and GUARD (Medicare Part D)—that would assess additional manufacturer rebates based on international benchmarks for certain therapeutic categories that include ophthalmology/ophthalmic agents. Because certain of our ophthalmic products are reimbursed under Medicare Part B, and we may develop or acquire additional products reimbursed by government programs, these and similar initiatives could be particularly relevant to our business. Moreover, because we do not own global rights to many of the products we market in the U.S. and generally do not control commercialization or pricing outside the U.S. for those products, we may have limited or no ability to affect non-U.S. pricing that could be used as a benchmark under most-favored-nation or international reference pricing initiatives, which could increase our exposure to such policies.
We cannot predict the scope, timing, or ultimate impact of these or other policy changes. However, to the extent payor actions or governmental measures decrease or modify coverage or reimbursement for our products, increase rebates, discounts, or other price concessions, shift additional costs to manufacturers, or otherwise limit utilization, our business, financial condition, results of operations, and cash flows could be materially adversely affected.
Changing U.S. federal coverage and reimbursement policies and practices have affected and are likely to continue to affect access to, pricing of and sales of our products.
A substantial portion of our branded product portfolio relies on reimbursement from federal government healthcare programs and commercial insurance plans regulated by federal and state governments. Our business has been and will continue to be affected by legislative actions changing U.S. federal reimbursement policy. The IRA’s drug pricing controls and Medicare redesign is likely to have a material adverse effect on our sales (particularly for our branded products that are more substantially reliant on Medicare reimbursement), our business and our results of operations. However, as the degree of impact from this legislation on our business depends on a number of implementation decisions, the extent of the IRA’s impact on our sales and, in turn, our business remains unclear.
| 29 |
Changing reimbursement and pricing actions in various states have negatively affected and may continue to negatively affect access to and have affected and may continue to affect sales of our products.
At the state level, government actions or ballot initiatives can also affect how our branded products are covered and reimbursed and/or create additional pressure on our pricing decisions. Existing and proposed state pricing laws have added complexity to the pricing of drugs and may already be affecting industry pricing decisions. A number of states have adopted, and many other states are considering, drug importation programs or other pricing actions, including proposals designed to require biopharmaceutical manufacturers to report to the state proprietary pricing information or provide advance notice of certain price increases. For example, California law requires biopharmaceutical manufacturers to notify health insurers and government health plans at least 60 days before scheduled prescription drug price increases that exceed certain thresholds. Similar laws exist in Oregon and Washington. Additional proposals directed at Medicaid seek to penalize manufacturers for pricing drugs above a certain threshold or limit spending on biopharmaceutical products. States are also seeking to change the way they pay for drugs for patients covered by state programs. New York has established a Medicaid drug spending cap, and Massachusetts implemented a new review and supplemental rebate negotiation process. Six states (Colorado, Maine, New Hampshire, Maryland, Oregon and Washington) have enacted laws that establish Prescription Drug Affordability Boards (“PDABs”) to study drug prices and identify drugs that pose affordability challenges, and in three states (Colorado, Maryland and Washington) include authority for the state PDABs to set upper payment limits on certain drugs in state regulated plans. Other states may consider implementing similar policies and laws. Additionally, Colorado, Florida, Maine, New Hampshire, New Mexico and Vermont have enacted laws, and several other states have proposed bills, to implement importation of drugs from Canada. The FDA has met with representatives from Colorado, Florida, Maine and New Mexico to discuss those states’ proposed importation programs, and the FDA may be working towards approving such plans. Other states could adopt similar approaches or could pursue different policy changes in a continuing effort to reduce their costs. Ultimately, as with U.S. federal government actions, existing or future state government actions or ballot initiatives may also have a material adverse effect on our product sales, business and results of operations.
U.S. commercial payor actions have affected and may continue to affect access to and sales of our products
Payors, including healthcare insurers, pharmacy benefit managers (“PBMs”), integrated healthcare delivery systems (vertically-integrated organizations built from consolidations of healthcare insurers and PBMs) and group purchasing organizations, increasingly seek ways to reduce their costs. With increasing frequency, payors are adopting benefit plan changes that shift a greater proportion of drug costs to patients. Such measures include more limited benefit plan designs, high deductible plans, higher patient co-pay or coinsurance obligations and more significant limitations on patients’ use of manufacturer commercial co-pay assistance programs. Further, government regulation of payors may affect these trends. For example, CMS finalized a policy for plan years starting on or after January 1, 2021 that has caused commercial payors to more widely adopt co-pay accumulator adjustment programs. Payors, including PBMs, have sought, and continue to seek, price discounts or rebates in connection with the placement of our branded products on their formularies or those they manage, and to also impose restrictions on access to or usage of our branded products (such as step therapy), require that patients receive the payor’s prior authorization before covering the product, and/or chosen to exclude certain indications for which our products are approved. In an effort to reduce barriers to access, we may reduce the net price of some of our branded products by providing greater discounts and rebates to payors (including PBMs that administer Medicare Part D prescription drug plans), and we may introduce a set of new National Drug Codes to make our branded products available at a lower list price. However, affordability of patient out-of-pocket co-pay cost has limited and may continue to limit patient use. Further, despite these net and list price reductions, some payors may restrict, patient access and may seek further discounts or rebates or take other actions, such as changing formulary coverage for some or all of our branded products. These factors have limited, and may continue to limit, patient affordability and use, negatively affecting sales of our branded products.
Further, significant consolidation in the health insurance industry has resulted in a few large insurers and PBMs, which places greater pressure on pricing and usage negotiations with biopharmaceutical manufacturers, significantly increasing discount and rebate requirements and limiting patient access and usage. For example, according to a 2024 press release from the Federal Trade Commission (“FTC”), the six largest PBMs manage nearly 95% of prescriptions filled in the United States. This high degree of consolidation among insurers and PBMs and other payors, including through integrated healthcare delivery systems and/or with specialty or mail-order pharmacies and pharmacy retailers, has increased the negotiating leverage such entities have over us and other biopharmaceutical manufacturers and has resulted in greater price discounts, rebates and service fees realized by those payors from our business. CVS, Express Scripts and United Health Group (among the top five integrated health plans and PBMs), each have Rebate Management Organizations that further increase their leverage to negotiate deeper discounts. Ultimately, additional discounts, rebates, fees, coverage changes, plan changes, restrictions or exclusions imposed by these commercial payors could have a material adverse effect on our product sales, business and results of operations. Policy reforms advanced by Congress or the others in the federal administration that refine the role of PBMs in the U.S. marketplace could have downstream implications or consequences for our business and how we interact with these entities.
| 30 |
Guidelines and recommendations published by various organizations can reduce the use of our branded products.
Government agencies promulgate regulations and guidelines directly applicable to us and to our products. Professional societies, practice management groups, insurance carriers, physicians’ groups, private health and science foundations and organizations involved in various diseases also publish guidelines and recommendations to healthcare providers, administrators and payors, as well as patient communities. Recommendations by government agencies or other groups and organizations may relate to such matters as usage, dosage, route of administration and use of related therapies. In addition, a growing number of organizations are providing assessments of the value and pricing of biopharmaceutical products, and even organizations whose guidelines have historically been focused on clinical matters have begun to incorporate analyses of the cost effectiveness of various treatments into their treatment guidelines and recommendations. Value assessments may come from private organizations that publish their findings and offer recommendations relating to the products’ reimbursement by government and private payors. Some companies and payors have announced pricing and payment decisions based in part on the assessments of private organizations. In addition, government health technology assessment organizations in many countries make reimbursement recommendations to payors in their jurisdictions based on the clinical effectiveness, cost-effectiveness and service effects of new, emerging and existing medicines and treatments. Such health technology assessment organizations have recommended, and may in the future recommend, reimbursement for certain of our products for a narrower indication than was approved by applicable regulatory agencies or may recommend against reimbursement entirely. See “Our sales depend on coverage and reimbursement from government and commercial third-party payors, and pricing and reimbursement pressures have affected, and are likely to continue to affect, our profitability”. Such recommendations or guidelines may affect our reputation, and any recommendations or guidelines that result in decreased use, dosage or reimbursement of our products could have a material adverse effect on our product sales, business and results of operations. In addition, the perception by the investment community or stockholders that such recommendations or guidelines will result in decreased use and dosage of our products could adversely affect the market price of our common stock.
Change in U.S. trade policy, including tariffs or other import restrictions, could increase our costs, disrupt our supply chain, and adversely affect our business.
U.S. trade policy is subject to change and could include the imposition of new or increased tariffs, duties, quotas, sanctions, or other restrictions on finished pharmaceuticals, APIs, excipients, packaging components, equipment, or other inputs used in our products. Although a significant portion of our products (compounded and branded) are manufactured in the United States, we and our third-party manufacturers and suppliers rely on global supply chains for certain finished pharmaceuticals APIs, materials, components, and services.
If tariffs or other trade restrictions are enacted or expanded, we could experience higher input and logistics costs, delays at ports of entry, reduced availability of materials or components, supplier disruptions, and increased working-capital requirements, including the need to build additional inventory or make advance payments of duties. In addition, changes in trade policy may prompt retaliatory actions by foreign governments or cause suppliers to reallocate capacity, which could further constrain supply or increase costs.
We may seek to mitigate these risks through supply chain planning and other measures, including identifying alternative suppliers, qualifying additional sources, modifying sourcing strategies, and adjusting inventory levels. However, there can be no assurance that these efforts will be successful, timely, or sufficient to offset increased costs or disruptions. Any sustained increase in costs or disruption in our supply chain could adversely affect our business, financial condition, results of operations, and cash flows.
| 31 |
Risks Related to Competition
There are many competitive risks related to marketing and selling our proprietary formulations and operating our compounding pharmacy business.
The pharmaceutical and pharmacy industries are highly competitive. We compete against branded drug companies, generic drug companies, outsourcing facilities and other compounding pharmacies. We are significantly smaller than some of our competitors. Currently we lack some of the financial and other resources needed to develop, produce, distribute and market our proprietary formulations at a level to capture a significant market share in these sectors. The drug products available through branded and generic drug companies with which our formulations compete have been approved for marketing and sale by the FDA and are required to be manufactured in facilities compliant with cGMP standards. Although we prepare our compounded formulations in accordance with the standards provided by USP 795 and USP 797 and applicable state and federal law, our proprietary compounded formulations are not required to be, and have not been, approved for marketing and sale by the FDA. As a result, some physicians may be unwilling to prescribe, and some patients may be unwilling to use, our formulations. Additionally, under federal and state laws applicable to our current compounding pharmacy operations, we are not permitted to prepare significant amounts of a specific formulation in advance of a prescription, compound quantities for office use or utilize a wholesaler for distribution of our formulations; instead, our compounded formulations must be prepared and dispensed in connection with a physician prescription for an individually identified patient. Pharmaceutical companies, on the other hand, are able to sell their FDA-approved products to large pharmaceutical wholesalers, which can in turn sell to and supply hospitals and retail pharmacies. Even if we are successful in registering certain of our facilities as outsourcing facilities, our business may not be scalable on the scope available to our competitors that produce FDA-approved drugs, which may limit our potential for profitable operations. These facets of our operations may subject our business to limitations our competitors with FDA-approved drugs may not face.
Our future success depends in large part on our ability to maintain a competitive position with respect to biotechnology and related pharmaceutical technologies.
Biotechnology and related pharmaceutical technologies have undergone and continue to be subject to rapid and significant change. Our future success will depend in large part on our ability to maintain a competitive position with respect to these technologies. Products developed by our competitors, including FDA-approved drugs and compounded formulations created by other pharmacies, could render our products and technologies obsolete or unable to compete. Any products that we develop may become obsolete before we recover expenses incurred in their development, which may require us to raise additional funds that may or may not be available. The competitive environment requires an ongoing, extensive search for medical and technological innovations and the ability to develop and market these innovations effectively, and we may not be competitive with respect to these factors. Other competitive factors include the safety and efficacy of a product, the size of the market for a product, the timing of market entry relative to competitive products, the availability of alternative compounded formulations or approved drugs, the price of a product relative to alternative products, the availability of third-party reimbursement, the success of sales and marketing efforts, brand recognition and the availability of scientific and technical information about a product. Although we believe we are positioned to compete favorably with respect to many of these factors, if our proprietary formulations are unable to compete with the products of our competitors, we may never gain market share or achieve sustained profitability.
Concentration of sales at certain of our wholesaler distributors and consolidation of private payors may negatively affect our business.
Certain of our distributors, customers and payors have substantial purchasing leverage, due to the volume of our products they purchase or the number of patient lives for which they provide coverage. The substantial majority of our U.S. branded product sales are made through four pharmaceutical product wholesaler distributors: McKesson Corporation, Cencora Inc., Western Wellness and Cardinal Health, Inc. These distributors, in turn, sell our products to their customers, which include physicians or their clinics, ambulatory surgical centers, hospitals and pharmacies. Similarly, as discussed above, there has been significant consolidation in the health insurance industry, including that a small number of PBMs now oversee a substantial percentage of total covered lives in the U.S. See “Our sales depend on coverage and reimbursement from government and commercial third-party payors, and pricing and reimbursement pressures have affected, and are likely to continue to affect, our profitability.” The three largest PBMs in the U.S. are now part of major health insurance providers. The growing concentration of purchasing and negotiating power by these entities has, and may continue to, put pressure on our pricing due to their ability to extract price discounts on our branded products, fees for other services or rebates, negatively affecting our bargaining position, sales and/or profit margins. In addition, decisions by these entities to purchase or cover less or none of our branded products in favor of competing products could have a material adverse effect on our branded product sales, business and results of operations due to their purchasing volume. Further, if one of our significant wholesale distributors encounters financial or other difficulties and becomes unable or unwilling to pay us all amounts that such distributor owes us on a timely basis, or at all, it could negatively affect our business and results of operations. In addition, if one of our significant wholesale distributors becomes insolvent or otherwise unable to continue its commercial relationship with us in its present form, it could significantly disrupt our business and adversely affect our product sales, our business and results of operations unless suitable alternatives are timely found or lost sales are absorbed by another distributor.
| 32 |
If we are unable to protect our proprietary rights, we may not be able to prevent others from using our intellectual property, which may reduce the competitiveness and value of the related assets.
Our success will depend in part on our ability to obtain and maintain patent protection for our formulations and technologies and to prevent third parties from infringing upon our proprietary rights. We must also operate without infringing upon patents and proprietary rights of others, including by obtaining appropriate licenses to patents or other proprietary rights held by third parties, if necessary. The primary means by which we will be able to protect our formulations and technologies from unauthorized use by third parties is to obtain valid and enforceable patents that cover them. However, the applications we have filed or may file in the future may never yield patents that protect our inventions and intellectual property assets. Failure to obtain patents that sufficiently cover our formulations and technologies would limit our protection against other compounding pharmacies and outsourcing facilities, generic drug manufacturers, pharmaceutical companies and other parties who may seek to copy our products, produce products substantially similar to ours or use technologies substantially similar to those we own. We have made, and expect to continue to make, significant investments in certain of our proprietary formulations prior to the grant of any patents covering these formulations, and we may not receive a sufficient return on these investments if patent coverage or other appropriate intellectual property protection is not obtained and their competitiveness and value decreases.
The patent and intellectual property positions of pharmacies and pharmaceutical companies, including ours, are uncertain and involve complex legal and factual questions. There is no guarantee that we have developed or obtained or will in the future develop or obtain the rights to products or processes that are patentable, that patents will issue from any pending applications or that claims allowed will be sufficient to protect the technology we have developed or may in the future develop or to which we have acquired or may in the future acquire development rights. In addition, we cannot be certain that patents issued to us will not be challenged, invalidated, infringed or circumvented, including by our competitors, or that the rights granted thereunder will provide competitive advantages to us. In certain instances, we have acquired products that are patented and have been subject to prior litigation challenging the validity of certain patents related to those products. In some situations, the litigation resulted in settlement agreements that have allowed generic manufacturers to license the patent rights related to certain products and allowing the generic manufacturer to enter the market prior to patent expiration associated with the branded product.
We also rely on unpatented trade secrets and know-how and continuing technological innovation in order to develop our products, formulations, which we seek to protect, in part, by confidentiality agreements with our employees, consultants, collaborators and others, including certain service providers. We also have invention or patent assignment agreements with our current employees and certain consultants. Nonetheless, our employees and consultants may breach these agreements, and we may not have adequate remedies for the breach. Our trade secrets may otherwise become known or be independently discovered by competitors or could be developed by a person not bound by an invention assignment agreement with us, in which case we may have no rights to use the applicable invention.
We may face additional competition outside of the U.S. as a result of a lack of patent coverage in some territories and differences in patent prosecution and enforcement laws in foreign counties.
Filing, prosecuting, defending and enforcing patents on our proprietary formulations throughout the world is extremely expensive. We do not currently have patent protection outside of the U.S. that covers any of our proprietary formulations or other assets that we are currently pursuing. Competitors may use our technologies to develop their own products in jurisdictions where we have not obtained patent protection.
Even if the international patent applications we have filed or may in the future file are issued or approved, it is likely that the scope of protection provided by such patents would be different from, and possibly less than, the scope provided by corresponding U.S. patents. As a result, patent rights we are able to obtain may not be sufficient to prevent generic competition. Further, the extent of our international market opportunity may be dependent upon the enforcement of patent rights in various other countries. A number of countries in which we could file patent applications have a history of weak enforcement and/or compulsory licensing of intellectual property rights. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which would make it difficult for us to stop a third party from infringing any of our intellectual property rights. Moreover, attempting to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business.
| 33 |
Our products, drug candidates and compounded formulations and technologies could potentially conflict with the rights of others.
The preparation or sale of our products, drug candidates and compounded formulations and use of our technologies may infringe on the patent or other intellectual property rights of others. If our products infringe or conflict with the patent or other intellectual property rights of others, third parties could bring legal actions against us claiming damages and seeking to enjoin our manufacturing and marketing of our affected products. Patent litigation is costly and time consuming and may divert management’s attention and our resources. We may not have sufficient resources to bring any actions to a successful conclusion. If we are not successful in defending against these legal actions should they arise, we may be subject to monetary liability or be forced to alter our products, cease some or all of our operations relating to the affected products, or seek to obtain a license in order to continue manufacturing and marketing the affected products, which may not be available on acceptable terms or at all.
We are dependent on our CEO, Mark L. Baum and President and CFO, Andrew R. Boll, for the continued growth and development of our Company.
Our CEO, Mark L. Baum and our President and CFO, Andrew R. Boll, have played a primary role in the founding of our business along with creating and developing our current business model. We are highly dependent on these executives for the implementation of our business plan and the future development of our assets and our business, and the loss of their services and leadership could materially adversely impact our Company.
If we are unable to attract and retain key personnel and consultants, we may be unable to maintain or expand our business.
We have been focusing on building our management, pharmacy, research and development, sales and marketing and other personnel to pursue our current business model. To achieve our planned growth, we may have significant difficulty attracting and retaining necessary employees. Because of the specialized nature of our business, our ability to develop products and to compete will remain highly dependent upon our ability to attract and retain qualified pharmacy, scientific, technical and commercial employees and consultants. There is intense competition to hire qualified personnel in our industry, and we may be unable to continue to attract and retain the qualified personnel necessary, particularly since our headquarters location is not near the primary centers of biopharmaceutical employment, for the development of our business. The loss of key employees or consultants or the failure to recruit or engage new employees and consultants could have a material adverse effect on our business. In addition, any staffing interruptions resulting from geopolitical actions, including war and terrorism, adverse public health developments, or natural disasters including earthquakes, typhoons, floods and fires, could have a material adverse effect on our business.
Risks Related to Product Development, Regulatory Approval, Manufacturing and Commercialization
If we seek FDA approval to market and sell any of our drug candidates we may be unable to demonstrate the necessary safety and efficacy to obtain such FDA approval.
In recent years, we have sought, and in the future, we, alone or with project partners, intend to seek, FDA regulatory approval to market and sell one or more of our assets as an FDA-approved drug. Obtaining FDA approval to market and sell pharmaceutical products is costly, time-consuming, uncertain and subject to unanticipated delays. The FDA or other regulatory agencies may not approve a drug candidate on a timely basis or at all. Before we obtain FDA approval for the sale of any potential drug candidates, we will be required to demonstrate through pre-clinical studies and clinical trials that it is safe and effective for each intended use, which we may not be able to do. A failure to demonstrate safety and efficacy of a drug candidate to the FDA’s satisfaction would result in our failure to obtain FDA approval. Moreover, even if the FDA were to grant regulatory approval of a drug candidate, the approval may be limited to specific therapeutic areas or limited as to its distribution, which could reduce revenue potential, and we will be subject to extensive and costly post-approval requirements and oversight with respect to commercialization of the drug candidate.
| 34 |
Even if we receive regulatory approval for any of our drug candidates, we may not be able to successfully commercialize the product and the revenue that we generate from its sales, if any, may be limited.
If approved for marketing, the commercial success of our drug candidates will depend upon each product’s acceptance by the medical community, including physicians, patients and health care payors. The degree of market acceptance for any of our drug candidates will depend on a number of factors, including:
| ● | demonstration of clinical safety and efficacy; | |
| ● | relative convenience, dosing burden and ease of administration; | |
| ● | the prevalence and severity of any adverse effects; | |
| ● | the willingness of physicians to prescribe our drug candidates, and the target patient population to try new therapies; | |
| ● | efficacy of our drug candidates compared to competing products; | |
| ● | the introduction of any new products that may in the future become available targeting indications for which our drug candidates may be approved; | |
| ● | new procedures or therapies that may reduce the incidences of any of the indications in which our drug candidates may show utility; | |
| ● | pricing and cost-effectiveness; | |
| ● | the inclusion or omission of our drug candidates in applicable therapeutic and vaccine guidelines; | |
| ● | the effectiveness of our own or any future collaborators’ sales and marketing strategies; | |
| ● | limitations or warnings contained in approved labeling from regulatory authorities; | |
| ● | our ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare and Medicaid, private health insurers and other third-party payors or to receive the necessary pricing approvals from government bodies regulating the pricing and usage of therapeutics; and | |
| ● | the willingness of patients to pay out-of-pocket in the absence of third-party coverage or reimbursement or government pricing approvals. |
If any of our drug candidates are approved, but do not achieve an adequate level of acceptance by physicians, health care payors, and patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate the medical community and third-party payors on the benefits of our drug candidates may require significant resources and may never be successful.
In addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize our drug candidates successfully. For example, if the approval process takes too long, we may miss market opportunities and give other companies the ability to develop competing products or establish market dominance. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render our drug candidates not commercially viable. For example, regulatory authorities may approve any of our drug candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for any of our drug candidates, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve any of our drug candidates with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that indication. Further, the FDA or comparable foreign regulatory authorities may place conditions on approvals or require risk management plans or a Risk Evaluation and Mitigation Strategy (“REMS”) to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA may also require a REMS for an approved product when new safety information emerges. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of our drug candidates. Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of our drug candidates.
| 35 |
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing of drug candidates is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of pre-clinical studies and early clinical trials may not be predictive of the results of later-stage clinical trials. We cannot assure you that the FDA or comparable foreign regulatory authorities will view the results as we do or that any future trials of any of our drug candidates will achieve positive results. Drug candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through pre-clinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Any future clinical trial results for our drug candidates may not be successful.
In addition, a number of factors could contribute to a lack of favorable safety and efficacy results for any of our drug candidates. For example, such trials could result in increased variability due to varying site characteristics, such as local standards of care, differences in evaluation period and surgical technique, and due to varying patient characteristics including demographic factors and health status.
Even if we obtain marketing approval for any of our drug candidates, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, our drug candidates could be subject to labeling and other restrictions and withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our drug candidates.
Even if we obtain regulatory approval for any of our drug candidates for an indication, the FDA or foreign equivalent may still impose significant restrictions on their indicated uses or marketing or the conditions of approval, or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials, and post-market surveillance to monitor safety and efficacy. Our drug candidates will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market information. These requirements include registration with the FDA, as well as continued compliance with current Good Clinical Practices regulations (“cGCPs”) for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current cGMPs, requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
The FDA has the authority to require a REMS as part of an NDA or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria or requiring patient testing, monitoring and/or enrollment in a registry.
With respect to sales and marketing activities by us or any future partner, advertising and promotional materials must comply with FDA rules in addition to other applicable federal, state and local laws in the U.S. and similar legal requirements in other countries. In the U.S., the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug Marketing Act. Application holders must obtain FDA approval for product and manufacturing changes, depending on the nature of the change. We may also be subject, directly or indirectly through our customers and partners, to various fraud and abuse laws, including, without limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act, and similar state laws, which impact, among other things, our proposed sales, marketing, and scientific/educational grant programs. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of the VA, or other government drug programs, we will be subject to complex laws and regulations regarding reporting and payment obligations. All of these activities are also potentially subject to U.S. federal and state consumer protection and unfair competition laws. Similar requirements exist in many of these areas in other countries.
| 36 |
In addition, if any of our drug candidates are approved for a particular indication, our product labeling, advertising and promotion would be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for our drug candidates, physicians may nevertheless legally prescribe our products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees of permanent injunctions under which specified promotional conduct is changed or curtailed.
If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, problems with the facility where the product is manufactured, or we or our manufacturers fail to comply with applicable regulatory requirements, we may be subject to the following administrative or judicial sanctions:
| ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; | |
| ● | issuance of warning letters or untitled letters; | |
| ● | clinical holds; | |
| ● | injunctions or the imposition of civil or criminal penalties or monetary fines; | |
| ● | suspension or withdrawal of regulatory approval; | |
| ● | suspension of any ongoing clinical trials; | |
| ● | refusal to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals; | |
| ● | suspension or imposition of restrictions on operations, including costly new manufacturing requirements; or | |
| ● | product seizure or detention or refusal to permit the import or export of product. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize our drug candidates and generate revenue. Adverse regulatory action, whether pre- or post-approval, can also potentially lead to product liability claims and increase our product liability exposure.
| 37 |
Delays in the completion of, or the termination of, any clinical or non-clinical trials for any drug candidates for which we may seek FDA approval could adversely affect our business.
Clinical trials are very expensive, time consuming, unpredictable and difficult to design and implement. The results of clinical trials may be unfavorable, they may continue for several years, and they may take significantly longer to complete and involve significantly more costs than expected. Delays in the commencement or completion of clinical testing could significantly affect product development costs and plans with respect to any drug candidate for which we seek FDA approval. The commencement and completion of clinical trials can be delayed and experience difficulties for a number of reasons, including delays and difficulties caused by circumstances over which we may have no control. For instance, approvals of the scope, design or trial site may not be obtained from the FDA and other required bodies in a timely manner or at all, agreements with acceptable terms may not be reached in a timely manner or at all with CROs to conduct the trials, a sufficient number of subjects may not be recruited and enrolled in the trials, and third-party manufacturers of the materials for use in the trials may encounter delays and problems in the manufacturing process, including failure to produce materials in sufficient quantities or of an acceptable quality to complete the trials. If we were to experience delays in the commencement or completion of, or if we were to terminate, any clinical or non-clinical trials we pursue in the future, the commercial prospects for the applicable drug candidates may be limited or eliminated, which may prevent us from recouping our investment in research and development efforts for the drug candidate and would have a material adverse effect on our business, results of operations, financial condition and prospects.
We may depend on the success of our drug candidates, which have not yet demonstrated efficacy for their target or any other indications. If we are unable to generate revenues from our drug candidates, our ability to create stockholder value may be limited.
Our drug candidates are in various stages of clinical development. There is no guarantee that our clinical trials will be successful or that we will continue clinical development in support of an approval from the FDA or comparable foreign regulatory authorities for any indication. We note that most drug candidates never reach the clinical development stage and even those that do commence clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval. Therefore, aspects of our business depend on the successful development, regulatory approval and commercialization of our drug candidates, which may never occur.
If we are not able to obtain required regulatory approvals for a drug candidate, we will not be able to commercialize such drug candidate and our ability to generate revenues will be limited.
We must successfully complete clinical trials for our drug candidates before we can apply for marketing approval. Even if we complete our clinical trials, it does not assure marketing approval. Our clinical trials may be unsuccessful, which would materially harm our business. Even if our initial clinical trials are successful, we are required to conduct additional clinical trials to establish our drug candidates’ safety and efficacy, before an NDA or Biologics License Application (“BLA”), or their foreign equivalents can be filed with the FDA or comparable foreign regulatory authorities for marketing approval of our drug candidates.
Clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in early phases of pre-clinical and clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our drug candidates. The research, testing, manufacturing, labeling, packaging, storage, approval, sale, marketing, advertising and promotion, pricing, export, import and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the U.S. and other countries, which regulations differ from country to country. We are not permitted to market our drug candidates as prescription pharmaceutical products in the U.S. until we receive approval of an NDA from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the U.S., the FDA generally requires the completion of clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are eventually approved for commercialization. If our development efforts for our drug candidates, including regulatory approval, are not successful for their planned indications, or if adequate demand for our drug candidates is not generated, our business will be materially adversely affected.
| 38 |
Our success depends on the receipt of regulatory approval and the issuance of such regulatory approvals is uncertain and subject to a number of risks, including the following:
| ● | the results of toxicology studies may not support the filing of an investigational new drug application for our drug candidates; | |
| ● | the FDA or comparable foreign regulatory authorities or Institutional Review Boards (“IRBs”) may disagree with the design or implementation of our clinical trials; | |
| ● | we may not be able to provide acceptable evidence of our drug candidates’ safety and efficacy; |
| ● | the results of our clinical trials may not be satisfactory or may not meet the level of statistical or clinical significance required by the FDA, the European Medicines Agency (the “EMA”), or other regulatory agencies for marketing approval; | |
| ● | the dosing of our drug candidates in a particular clinical trial may not be at an optimal level; | |
| ● | patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to our drug candidates; | |
| ● | the data collected from clinical trials may not be sufficient to support the submission of an NDA, BLA or other submission or to obtain regulatory approval in the U.S. or elsewhere; | |
| ● | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and | |
| ● | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
Failure to obtain regulatory approval for our drug candidates for the foregoing, or any other reasons, will prevent us from commercializing our drug candidates, and our ability to generate revenue will be materially impaired. We cannot guarantee that regulators will agree with our assessment of the results of the clinical trials we intend to conduct in the future or that such trials will be successful. The FDA, EMA and other regulators have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional clinical trials, or pre-clinical or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of our drug candidates.
Excluding any activities through our prior ownership interest in Eton, we have not received regulatory approval to market our drug candidates in any jurisdiction. We have only limited experience in filing the applications necessary to gain regulatory approvals and expect to rely on consultants and CROs, with expertise in this area to assist us in this process. Securing regulatory approvals to market a product requires the submission of pre-clinical, clinical, and/or pharmacokinetic data, information about product manufacturing processes and inspection of facilities and supporting information to the appropriate regulatory authorities for each therapeutic indication to establish a drug candidate’s safety and efficacy for each indication. Our drug candidates may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude us from obtaining regulatory approval or prevent or limit commercial use with respect to one or all intended indications.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon, among other things, the type, complexity and novelty of the drug candidates involved, the jurisdiction in which regulatory approval is sought and the substantial discretion of the regulatory authorities. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for a submitted product application may cause delays in the approval or rejection of an application. Regulatory approval obtained in one jurisdiction does not necessarily mean that a drug candidate will receive regulatory approval in all jurisdictions in which we may seek approval, but the failure to obtain approval in one jurisdiction may negatively impact our ability to seek approval in a different jurisdiction. Failure to obtain regulatory marketing approval for our drug candidates in any indication will prevent us from commercializing the drug candidate, and our ability to generate revenue will be materially impaired.
| 39 |
Obtaining and maintaining regulatory approval of our products and drug candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our products or drug candidates in other jurisdictions.
Obtaining and maintaining regulatory approval of our drug candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a drug candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the drug candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the U.S., including additional pre-clinical studies or clinical trials, as clinical studies conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the U.S., a drug candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. If we fail to comply with the regulatory requirements in international markets and/ or to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our drug candidates will be harmed.
Current and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our drug candidates and affect the prices we may obtain.
In the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for our drug candidates, restrict or regulate post-approval activities and affect our ability to profitably sell our drug candidates. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our drug candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In the U.S., the Medicare Modernization Act (the “MMA”) changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for our drug candidates and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
The Health Care Reform Law is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. Further, the law imposed a significant annual fee on companies that manufacture or import branded prescription drug products.
| 40 |
The Health Care Reform Law remains subject to legislative efforts to repeal, modify or delay the implementation of the law. Efforts to date have generally been unsuccessful. If the Health Care Reform Law is repealed or modified, or if implementation of certain aspects of the Health Care Reform Law are delayed, such repeal, modification or delay may materially adversely impact our business, strategies, prospects, operating results or financial condition. We are unable to predict the full impact of any repeal or modification in the implementation of the Health Care Reform Law on us at this time.
In addition, other legislative changes have been proposed and adopted in the U.S. since the Health Care Reform Law was enacted. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce or eliminate our profitability.
Our drug candidates may face competition sooner than expected.
Our success will depend in part on our ability to obtain and maintain patent protection for certain of our drug candidates and technologies and to prevent third parties from infringing upon our proprietary rights. We must also operate without infringing upon patents and proprietary rights of others, including by obtaining appropriate licenses to patents or other proprietary rights held by third parties, if necessary. However, the applications we have filed or may file in the future may never yield patents that protect our inventions and intellectual property assets. Failure to obtain patents that sufficiently cover our formulations and technologies would limit our protection against compounding pharmacies, outsourcing facilities, generic drug manufacturers, pharmaceutical companies and other parties who may seek to copy our products, produce products substantially similar to ours or use technologies substantially similar to those we own.
We also intend to seek data exclusivity or market exclusivity for our drug candidates provided under the FDCA and similar laws in other countries. The FDCA provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving NDAs for drugs containing the original active agent. Even if our drug candidates are considered to be reference products eligible for three years of exclusivity under the FDCA, another company could market competing products if the FDA approves a full NDA for such product containing the sponsor’s own pre-clinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products. Moreover, an amendment or repeal of the FDCA could result in a shorter exclusivity period for our drug candidates, which would have a material adverse effect on our business.
We are and will be completely dependent on third parties to manufacture our branded drug products and drug candidates, and our commercialization of our drug candidates could be halted, delayed or made less profitable if those third parties fail to obtain manufacturing approval from the FDA or comparable foreign regulatory authorities, fail to provide us with sufficient quantities of our drug candidates or fail to do so at acceptable quality levels or prices.
We do not currently have, nor do we plan to acquire, the capability or infrastructure to manufacture the API in our drug candidates for use in our clinical trials or for commercial product. In addition, we do not have the capability to manufacture any of our branded drug products and candidates as a finished drug product for commercial distribution. As a result, we are and will be obligated to rely on contract manufacturers.
| 41 |
The facilities used by our contract manufacturers to manufacture our drug products and candidates must be approved by the FDA or comparable foreign regulatory authorities pursuant to inspections that will be conducted after we submit an NDA or BLA to the FDA or their equivalents to other relevant regulatory authorities. We will not control the manufacturing process of, and will be completely dependent on, our contract manufacturing partners for compliance with cGMPs for manufacture of both active drug substances and finished drug products. These cGMP regulations cover all aspects of the manufacturing, testing, quality control and record keeping relating to our drug candidates. If our contract manufacturers do not successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our drug candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our drug candidates, if approved.
Our contract manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. We do not have control over our contract manufacturers’ compliance with these regulations and standards. Failure by any of our contract manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure to grant approval to market any of our drug candidates, delays, suspensions or withdrawals of approvals, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business. In addition, we do not have control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. Failure by our contract manufacturers to comply with or maintain any of these standards could adversely affect our ability to develop, obtain and maintain regulatory approval for or market any of our drug products and drug candidates.
If, for any reason, these third parties are unable or unwilling to perform, we may not be able to terminate our agreements with them, and we may not be able to locate alternative manufacturers or formulators or enter into favorable agreements with them, and we cannot be certain that any such third parties will have the manufacturing capacity to meet future requirements. If these manufacturers or any alternate manufacturer of finished drug product experiences any significant difficulties in its respective manufacturing processes for our API or finished products or should cease doing business with us, we could experience significant interruptions in the supply of any of our drug candidates or may not be able to create a supply of our drug candidates at all. Were we to encounter manufacturing issues, our ability to produce a sufficient supply of any of our drug candidates might be negatively affected. Our inability to coordinate the efforts of our third-party manufacturing partners, or the lack of capacity available at our third-party manufacturing partners, could impair our ability to supply any of our drug candidates at required levels. Because of the significant regulatory requirements that we would need to satisfy in order to qualify a new bulk or finished product manufacturer, if we face these or other difficulties with our current manufacturing partners, we could experience significant interruptions in the supply of any of our drug candidates if we decided to transfer the manufacture of any of our drug candidates to one or more alternative manufacturers in an effort to deal with the difficulties.
Any manufacturing problem or the loss of a contract manufacturer could be disruptive to our operations and result in lost sales. Additionally, we rely on third parties to supply the raw materials needed to manufacture our existing and potential products. Any business interruptions resulting from geopolitical actions, including war and terrorism, adverse public health developments, or natural disasters including earthquakes, typhoons, floods and fires, could affect our supply chain. Any reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to a future contract manufacturer caused by problems at suppliers could delay shipment of any of our drug candidates, increase our cost of goods sold and result in lost sales.
We expect to rely on third parties to conduct clinical trials for our drug candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize any of our drug candidates, and our business would be substantially harmed.
We expect to enter into agreements with third-party CROs to conduct and manage our clinical programs, including contracting with clinical sites to perform our clinical studies. We plan to rely heavily on these parties for execution of clinical studies for our drug candidates and will control only certain aspects of their activities. Nevertheless, we will be responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on CROs and clinical sites will not relieve us of our regulatory responsibilities. We and our CROs will be required to comply with cGCPs, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for any products in clinical development. The FDA and its foreign equivalents enforce these cGCP regulations through periodic inspections of trial sponsors, principal investigators and trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA or other regulatory authorities will determine that any of our clinical trials comply with cGCPs. In addition, our clinical trials must be conducted with products produced under cGMP regulations and will require a large number of test subjects. Our failure or the failure of our CROs or clinical sites to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
| 42 |
Although we intend to design the clinical trials for our drug candidates in consultation with CROs, we expect that the CROs will manage all of the clinical trials conducted at contracted clinical sites. As a result, many important aspects of our drug development programs would be outside of our direct control. In addition, the CROs and clinical sites may not perform all of their obligations under arrangements with us or in compliance with regulatory requirements. If the CROs or clinical sites do not perform clinical trials in a satisfactory manner, breach their obligations to us or fail to comply with regulatory requirements, the development and commercialization of any of our drug candidates for the subject indication may be delayed or our development program materially and irreversibly harmed. We cannot control the amount and timing of resources these CROs and clinical sites will devote to our program or any of our drug candidates. If we are unable to rely on clinical data collected by our CROs, we could be required to repeat, extend the duration of, or increase the size of our clinical trials, which could significantly delay commercialization and require significantly greater expenditures.
If any of our relationships with these third-party CROs or clinical sites terminate, we may not be able to enter into arrangements with alternative CROs or clinical sites. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any such clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our drug candidates. As a result, our financial results and the commercial prospects for any of our drug candidates would be harmed, our costs could increase and our ability to generate revenue could be delayed.
Any termination or suspension of, or delays in the commencement or completion of, any necessary studies of any of our drug candidates for any indications could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects.
The commencement and completion of clinical studies can be delayed for a number of reasons, including delays related to:
| ● | the FDA or a comparable foreign regulatory authority failing to grant permission to proceed and placing the clinical study on hold; | |
| ● | subjects for clinical testing failing to enroll or remain in our trials at the rate we expect; | |
| ● | a facility manufacturing any of our drug candidates being ordered by the FDA or other government or regulatory authorities to temporarily or permanently shut down due to violations of cGMP requirements or other applicable requirements, or cross-contaminations of drug candidates in the manufacturing process; | |
| ● | any changes to our manufacturing process that may be necessary or desired; | |
| ● | subjects choosing an alternative treatment for the indications for which we are developing our drug candidates, or participating in competing clinical studies; | |
| ● | subjects experiencing severe or unexpected drug-related adverse effects; | |
| ● | reports from clinical testing on similar technologies and products raising safety and/or efficacy concerns; | |
| ● | third-party clinical investigators losing their license or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or employing methods consistent with the clinical trial protocol, cGMP requirements, or other third parties not performing data collection and analysis in a timely or accurate manner; | |
| ● | inspections of clinical study sites by the FDA, comparable foreign regulatory authorities, or Institutional Review Boards (“IRBs”) finding regulatory violations that require us to undertake corrective action, result in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study, or that prohibit us from using some or all of the data in support of our marketing applications; |
| 43 |
| ● | third-party contractors becoming debarred or suspended or otherwise penalized by the FDA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or any of the data produced by such contractors in support of our marketing applications; | |
| ● | one or more IRBs refusing to approve, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | deviations of the clinical sites from trial protocols or dropping out of a trial; | |
| ● | adding new clinical trial sites; | |
| ● | the inability of the CRO to execute any clinical trials for any reason; and | |
| ● | government or regulatory delays or “clinical holds” requiring suspension or termination of a trial. |
Product development costs for any of our drug candidates will increase if we have delays in testing or approval or if we need to perform more or larger clinical studies than planned. Additionally, changes in regulatory requirements and policies may occur and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to the FDA, comparable foreign regulatory authorities, and IRBs for reexamination, which may impact the costs, timing or successful completion of that study. If we experience delays in completion of, or if we, the FDA or other regulatory authorities, the IRB, or other reviewing entities, or any of our clinical study sites suspend or terminate any of our clinical studies of any of our drug candidates, its commercial prospects may be materially harmed and our ability to generate product revenues will be delayed. Any delays in completing our clinical trials will increase our costs, slow down our development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical studies may also ultimately lead to the denial of regulatory approval of our drug candidates. In addition, if one or more clinical studies are delayed, our competitors may be able to bring products to market before we do, and the commercial viability of any of our drug candidates could be significantly reduced.
Even though we may apply for orphan drug designation for a drug candidate, we may not be able to obtain orphan drug marketing exclusivity.
There is no guarantee that the FDA, EMA or their foreign equivalents will grant any future application for orphan drug designation for any of our drug candidates, which would make us ineligible for the additional exclusivity and other benefits of orphan drug designation.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making a drug available in the Unites States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of regulatory review and approval process. In addition to the potential period of exclusivity, orphan designation makes a company eligible for grant funding of up to $650,000 per year for four years to defray costs of clinical trial expenses, tax credits for clinical research expenses and potential exemption from the FDA application user fee.
| 44 |
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. There can be no assurance that we will receive orphan drug designation for any of our drug candidates in the indications for which we think they might qualify, if we elect to seek such applications.
Although we may pursue expedited regulatory approval pathways for a drug candidate, it may not qualify for expedited development or, if it does qualify for expedited development, it may not actually lead to a faster development or regulatory review or approval process.
Although we believe there may be an opportunity to accelerate the development of certain of our drug candidates through one or more of the FDA’s expedited programs, such as fast track, breakthrough therapy, accelerated approval or priority review, we cannot be assured that any of our drug candidates will qualify for such programs.
For example, a drug may be eligible for designation as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Although breakthrough designation or access to any other expedited program may expedite the development or approval process, it does not change the standards for approval. If we apply for breakthrough therapy designation or any other expedited program for our drug candidates, the FDA may determine that our proposed target indication or other aspects of our clinical development plans do not qualify for such expedited program. Even if we are successful in obtaining a breakthrough therapy designation or access to any other expedited program, we may not experience faster development timelines or achieve faster review or approval compared to conventional FDA procedures. Access to an expedited program may also be withdrawn by the FDA if it believes that the designation is no longer supported by data from our clinical development program. Additionally, qualification for any expedited review procedure does not ensure that we will ultimately obtain regulatory approval for such drug candidate.
Disruptions at the FDA, CMS, the SEC, and other government agencies—due to funding lapses, shutdowns, staffing constraints, reorganizations, or shifting policy priorities—could adversely affect our business and results of operations.
Our business depends in part on timely and predictable interactions with U.S. federal agencies, including the FDA, CMS, and the SEC. Periodic disruptions to agency operations—whether caused by lapses in appropriations or government shutdowns, reduced staffing levels, reorganizations, budget constraints, or shifts in policy priorities—may delay or limit an agency’s ability to perform its customary functions. The federal government has experienced recent funding-related disruptions, and future disruptions remain possible.
Such disruptions could, among other things, delay FDA review of submissions, scheduling or completion of inspections, issuance of guidance, or other regulatory actions; delay CMS coverage, reimbursement, or payment-policy activities; or constrain the SEC’s ability to review filings and registration statements and to conduct other oversight activities. During funding lapses, agency operations may be significantly curtailed under contingency plans, which can contribute to backlogs and extended timelines. In addition, announced federal reorganizations and workforce reductions affecting the U.S. Department of Health and Human Services and certain of its operating divisions, including the FDA and CMS, may create further uncertainty regarding agency capacity, timing, and priorities.
| 45 |
If agency capacity is reduced or agency priorities shift, we could experience greater regulatory uncertainty, slower review and decision timelines, increased compliance costs, and delays in executing aspects of our strategy. Further, funding lapses or broader governmental disruption could contribute to volatility in financial markets and reduced liquidity, which could adversely affect our ability to access capital on acceptable terms.
Risks Related to Our Indebtedness
We have incurred significant indebtedness, which will require substantial cash to service and which subjects us to certain financial requirements and business restrictions.
In September 2025, we issued $250 million aggregate principal amount of 8.625% Senior Notes due September 2030 (the “2030 Notes”) and used the proceeds to refinance debt. All our prior debt was repaid in full. We may incur additional indebtedness in the future. Our ability to make scheduled payments on our indebtedness depends on our future performance and ability to raise additional capital, which is subject to economic, financial, competitive and other factors, some of which are beyond our control. If we are unable to generate sufficient cash to service our debt, we may be required to adopt one or more alternatives, such as selling assets, restructuring our debt or obtaining additional capital through equity sales or incurrence of additional debt on terms that may be onerous or highly dilutive to our stockholders. Our ability to engage in any of these activities would depend on the capital markets and our financial condition at such time, and we may not be able to do so when needed, on desirable terms or at all, which could result in a default on our debt obligations. Additionally, our debt instruments contain, or from time to time may contain, various restrictive covenants, including, among others, our obligation to deliver certain financial and other information, our obligation to comply with certain notice and insurance requirements, and our inability, without prior consent, to dispose of certain of our assets, incur certain additional indebtedness, enter into certain merger, acquisition or change of control transactions, pay certain dividends or distributions on or repurchase any of our capital stock or incur any lien or other encumbrance on our assets, subject to certain permitted exceptions. Any failure by us to comply with any of these covenants, subject to certain cure periods, or to make all payments under the debt instruments when due, would cause us to be in default under the applicable debt instrument. In the event of any such default, lenders may be able to foreclose on our assets that secure the debt or declare all borrowed funds, together with accrued and unpaid interest, immediately due and payable, thereby potentially causing all of our available cash to be used to pay our indebtedness or forcing us into bankruptcy or liquidation if we do not then have sufficient cash available. Any such event or occurrence could severely and negatively impact our operations and prospects.
We could enter into various transactions that could increase the amount of our outstanding debt or adversely affect our capital structure or credit rating.
Subject to certain limited exceptions, the terms of the 2030 Notes do not prevent us from entering into a variety of acquisition, divestiture, refinancing, recapitalization or other highly leveraged transactions. As a result, we could enter into any such transaction even though the transaction could increase the total amount of our outstanding indebtedness, adversely affect our capital structure or credit rating or otherwise adversely affect the holders of the 2030 Notes.
Risks Related to Our Common Stock
If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results, which could cause our stock price to fall.
Effective internal controls are necessary for us to provide reliable financial results. If we cannot provide reliable financial results, our consolidated financial statements could be misstated, our reputation may be harmed and the trading price of our common stock could decline. As we discuss in Item 9A of this Annual Report, our management concluded that our internal controls over financial reporting were effective as of December 31, 2025. However, our controls over financial processes and reporting may not continue to be effective or we may identify material weaknesses or significant deficiencies in our internal controls in the future. Any failure to remediate any future material weaknesses or successfully implement required new or improved controls, could harm our operating results, cause us to fail to meet our reporting obligations or result in material misstatements in our consolidated financial statements or other public disclosures. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
| 46 |
Our stock price may be volatile.
The market price of our common stock has been and is likely to continue to be highly volatile. The market price could fluctuate widely in response to various factors, many of which are beyond our control, including our ability to execute our business plan; operating results that fall below expectations; industry or regulatory developments; investor perception of our industry or our prospects; economic and other external factors; and the other risk factors discussed in this “Risk Factors” section.
In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of our common stock.
We have the right to issue shares of preferred stock without obtaining stockholder approval. If we were to issue preferred stock, it may have rights, preferences and privileges superior to those of our common stock.
We are authorized to issue 5,000,000 shares of “blank check” preferred stock, with such rights, preferences and privileges as may be determined from time to time by our Board of Directors. Our Board of Directors is empowered, without stockholder approval, to issue preferred stock at any time in one or more series and to fix the dividend rights, dissolution or liquidation preferences, redemption prices, conversion rights, voting rights and other rights, preferences and privileges for any series of our preferred stock that may be issued. The issuance of shares of preferred stock, depending on the rights, preferences and privileges attributable to the preferred stock, could reduce the voting rights and powers of our common stockholders and the portion of our assets allocated for distribution to our common stockholders in a liquidation event, and could also result in dilution to the book value per share of our common stock. The preferred stock could also be utilized, under certain circumstances, as a method for raising additional capital or discouraging, delaying or preventing a change in control of our Company.
We have not paid dividends in the past and do not expect to pay dividends in the future. Any return on an investment will be limited to any appreciation in the value of our common stock.
We have never paid cash dividends on our common stock and do not anticipate doing so in the foreseeable future. Any payment of dividends on our common stock would depend on contractual restrictions, as well as our earnings, financial condition and other business and economic factors as our Board of Directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if our stock price appreciates.
Offers or availability for sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
The sale of substantial amounts of our common stock in the public market, or the perception that sales could occur, may cause the market price of our common stock to fall. Sales could occur upon the expiration of any statutory holding period, such as under Rule 144 under the Securities Act of 1933, as amended, applicable to outstanding shares, upon expiration of any lock-up periods applicable to outstanding shares, upon our issuance of shares upon the exercise of outstanding options or warrants, or upon our issuance of shares pursuant offerings of our equity securities. The availability for sale of a substantial number of shares of our common stock, whether or not sales have occurred or are occurring, also could make it more difficult for us to raise additional financing through the sale of equity or equity-related securities in the future, when needed, on acceptable terms or at all.
| 47 |
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price.
From time to time, global credit and financial markets have experienced extreme volatility and disruptions, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment and continued unpredictable and unstable market conditions. If the equity and credit markets deteriorate, it may make any debt or equity financing more difficult to complete, more costly, and more dilutive. In the event the Company or one of its subsidiaries needed to access additional capital, failure to secure financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive an economic downturn, which could directly affect our ability to attain our operating goals on schedule and on budget.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
We
utilize a layered approach in assessing, identifying, evaluating and managing material risks from cybersecurity threats, and leverage
outside partners to gain intelligence on threats. We take input from industry activities, third party assessments and internal simulations
and continuously adjust our protection mechanisms to be effective. We also assess operational and data security risks associated with
our use of third-party service providers, understanding where failure points may exist within our supply chain operations and data protections.
If we learn of a cybersecurity incident at a
| 48 |
We
face a number of cybersecurity risks in connection with our business. Although we have numerous controls to protect against common attacks,
some attacks may still be effective. Our controls are designed to detect, triage and eradicate these attacks. While we carry a cyber
insurance policy to help cover investigation and mitigation expenses, it may be subject to limitations and be insufficient to cover all
expenses that may result from a cybersecurity incident.
For more information about the cybersecurity risks and other information technology and data privacy risks we face, see Item 1A. “Risk Factors including the risk factor titled “A breakdown of our information technology systems, or a cyberattack or information security breach could significantly compromise the confidentiality, integrity and availability of our information technology systems, network-connected control systems and/or our data, interrupt the operation of our business and/or affect our reputation.”
ITEM 2. PROPERTIES
We lease approximately 17,700 square feet of office space in Nashville, Tennessee for our corporate headquarters. The current lease term expires on June 30, 2032 and includes the option to extend the term for two additional, consecutive five-year terms. This office serves as our corporate headquarters.
We lease approximately 11,600 square feet of lab and other office space in Nashville, Tennessee. The current lease term commenced in June 2022 and expires in June 2027. This office generally serves as ImprimisRx’s customer service center and analytical laboratory.
We lease approximately 38,200 square feet of lab, warehouse, and office space in Ledgewood, New Jersey, in three separate suites. The current lease term expires on July 31, 2027 and includes options to extend the lease term through 2037. This space serves as an outsourcing facility and pharmacy for ImprimisRx.
ITEM 3. LEGAL PROCEEDINGS
See Note 18 to our consolidated financial statements included in this Annual Report for information on various legal proceedings, which is incorporated into this Item by reference.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 49 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information for Common Stock
Our common stock is listed on The Nasdaq Stock Market LLC under the symbol “HROW”.
Performance Graph
Our common stock is traded on The Nasdaq Stock Market LLC and is a component of both the Nasdaq Composite Index and the Nasdaq Biotechnology Index. The following graph shows cumulative total stockholder return (i.e., price change plus reinvestment of dividends) on our common stock over the period commencing December 31, 2020 and ending on December 31, 2025, to that of the total return for the Nasdaq Composite Index and the Nasdaq Biotechnology Index, assuming an investment of $100 on December 31, 2020. The stock performance shown on the graph represents historical stock performance and is not necessarily indicative of future stock price performance
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Harrow Inc., the NASDAQ Composite Index and the NASDAQ Biotechnology Index
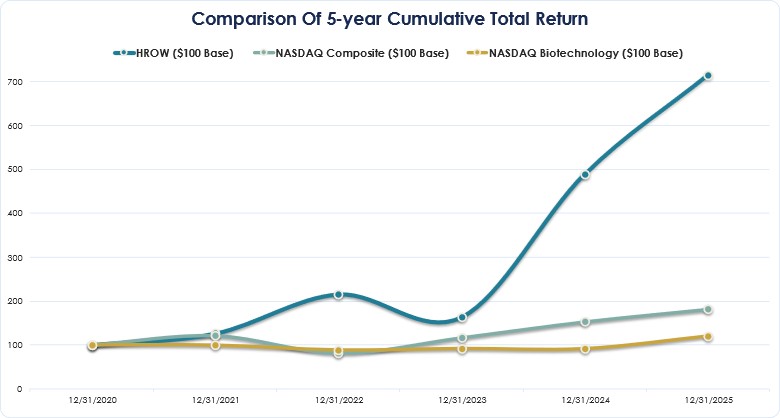
*$100 Invested on 12/31/2020 in stock or index, including reinvestment of dividends.
Fiscal year ending December 31.
The graph and related information is furnished under this Part II, Item 5 of this Annual Report on Form 10-K and shall not be deemed “soliciting material” or be “filed” with the SEC and shall not be incorporated by reference into any filing we make under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation by reference language in such filing.
Holders
As of February 25, 2026, there were approximately 56 stockholders of record (excluding an indeterminable number of stockholders whose shares are held in street or “nominee” name) of our common stock.
Dividends
We have not paid any dividends on our common stock since our inception and do not expect to pay dividends on our common stock in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plans
The information required by this item will be set forth in the definitive proxy statement we will file in connection with our 2026 Annual Meeting of Stockholders and is incorporated by reference herein.
| 50 |
Issuer Purchases of Equity Securities
During the three months ended December 31, 2025, the Company withheld shares of common stock to satisfy employee minimum statutory tax withholding obligations payable upon the vesting of restricted stock, as follows:
| Period | Total Number of Shares Purchased (a) | Weighted Average Price Paid per Share | Total Number of Shares Part of Publicly Announced Plans | Maximum Number of Shares Under the Plans or Programs (b) | ||||||||||||
| October1 – October 31 | 631 | 39.36 | - | - | ||||||||||||
| November 1 – November 30 | 73 | 37.77 | - | - | ||||||||||||
| December 1 – December 31 | - | $ | - | - | - | |||||||||||
| Total | 704 | $ | 39.20 | - | - | |||||||||||
(a) |
Represents shares withheld to satisfy the payment of tax obligations related to the vesting of restricted stock awards. |
(b) |
We had no publicly announced repurchase programs for shares of our common stock during the year ended December 31, 2025. |
Recent Sales of Unregistered Securities
None.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with the consolidated financial statements and the related notes contained in this Annual Report on Form 10-K (this “Annual Report”). Our consolidated financial statements have been prepared and, unless otherwise stated, the information derived therefrom as presented in this discussion and analysis is presented, in accordance with accounting principles generally accepted in the U.S. (GAAP). In addition to historical information, the following discussion contains forward-looking statements based upon our current views, expectations and assumptions that are subject to risks and uncertainties. Actual results may differ substantially from those expressed or implied by any forward-looking statements due to a number of factors, including, among others, the risks described in the “Risk Factors” section and elsewhere in this Annual Report. Additional information related to the comparison of our results of operations and liquidity and capital resources between the years 2024 and 2023 is included in Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations of our 2024 Form 10-K filed with the SEC and is incorporated by reference herin.
As used in this discussion and analysis, unless the context indicates otherwise, the terms the “Company,” “Harrow” “we,” “us” and “our” refer to Harrow, Inc. and its consolidated subsidiaries, including Imprimis RxNJ, LLC, Imprimis NJOF, LLC, ImprimisRx, LLC, Harrow IP, LLC and Harrow Eye, LLC.
| 51 |
Overview
We are a leading provider of ophthalmic disease management solutions in North America, and were founded with a commitment to deliver safe, effective, accessible, and affordable medications that enhance patient compliance and improve clinical outcomes. For over a decade, we have partnered with U.S. eyecare professionals to develop a comprehensive portfolio of high-quality products used to manage ophthalmic conditions affecting both the front and back of the eye, such as dry eye disease, wet (or neovascular) age-related macular degeneration, cataracts, refractive errors, glaucoma, and a range of other ocular surface conditions and retina diseases. By prioritizing clinical value – to the provider and the patient – Harrow empowers professionals to enhance patient outcomes and preserve vision. By combining our culture of creativity, entrepreneurship and groundbreaking innovation with operational discipline and strong financial performance, we are building a future where life-changing ophthalmic treatments are within reach for all.
Factors Affecting Our Performance
We believe the primary factors affecting our performance are our ability to increase revenues of our branded pharmaceutical products, grow and gain operating efficiencies in our operations, avoid or mitigate any potential regulatory-related restrictions, optimize pricing and obtain reimbursement options for our drug products, and continue to pursue development and commercialization opportunities for certain assets that we have not yet made commercially available. We believe we have built a tangible and intangible infrastructure that will allow us to scale revenues efficiently in the near and long-term. All of these activities will require significant costs and other resources, which we may not have or be able to obtain from operations or other sources. See “Liquidity and Capital Resources” below.
Recent Developments
The following describes certain developments in 2025 and 2026 to date that are important to understand our financial condition, results of operations, and expectations. See the notes to our consolidated financial statements included in this Annual Report for additional information about certain developments.
Commercial and Sales Force Expansions
In February 2026, we announced several commercial investments expected to be implemented during 2026 to support growth across key products. For VEVYE, following recent payor-coverage wins effective January 1, 2026, we began recruiting efforts to expand our commercial sales team from approximately 50 to 100 U.S. sales territories by late May 2026. For IHEEZO, we have begun expanding our commercial focus beyond retina practices into office-based ophthalmic procedures, targeting a broader set of anesthesia-dependent, reimbursed use cases, including non-retina intravitreal and subconjunctival injections, YAG/laser procedures, foreign body removals, and selected ocular surface and eyelid procedures. For TRIESENCE, we expect to increase the size of our dedicated sales force for this product during the coming months in response to favorable surgeon feedback and improving demand indicators, including increased interest in adoption and reordering for on-label uses in both office and surgical settings.
Acquisition of Remaining Interests in Melt Pharmaceuticals, Inc.
In September 2025, we entered into the Merger Agreement by and among Harrow, Harrow Acquisition Sub, Inc., a wholly owned subsidiary of Harrow, Melt, and D. Brad Osborne, as stockholder representative. Under the terms of the Merger Agreement and a related milestone payment agreement, we agreed to acquire the remaining equity interests of Melt in exchange for an initial cash payment of approximately $4,300,000 at closing, and contingent consideration consisting of cash and Company equity upon achievement of (i) FDA approval of the MELT-300 drug candidate, (ii) coding and reimbursement of the MELT-300 drug candidate, and (iii) various one-time sales milestones, as follows:
|
● |
Upon FDA approval of MELT-300, we shall pay an aggregate amount in cash of approximately $87,200,000. |
|
|
|
|
● |
Upon receipt of pass-through status awarded and J-Code (or any other similar designation) issued by CMS for MELT-300, we shall issue an aggregate of approximately 1,112,000 shares of our common stock. |
|
|
|
|
● |
Upon achievement of various annual net sales milestones ranging from $100,000,000 to $1,000,000,000 per year, we shall make various one-time cash payments that in the aggregate total up to approximately $260,000,000 if all annual net sales milestones are achieved. |
| 52 |
The regulatory and commercial milestones must be achieved, if at all, on or before December 31, 2035.
The Melt acquisition closed on November 17, 2025, and was treated as an asset acquisition for accounting purposes. As a result of such transaction, Melt’s drug candidates are now owned by Harrow and its R&D activities subsequent to the acquisition are included in Harrow’s consolidated financial results as of the year ended December 31, 2025.
Fifth Third Revolving Credit Facility
In September 2025, we entered into a Credit Agreement (the “5/3 Revolver”) with Fifth Third Bank, National Association, as administrative agent for itself and the other lenders (collectively, “Fifth Third”) providing for a senior secured revolving credit facility in the initial principal amount of $40,000,000, together with an uncommitted incremental revolving line of credit in the principal amount of up to $20,000,000. The 5/3 Revolver will mature on September 26, 2030, or, if earlier, the date that is 91 days prior to the earliest maturity date of the Company’s 2030 Notes.
Borrowings under the 5/3 Revolver bear interest at a floating rate equal to, at the Company’s option, either (i) a base rate plus a margin ranging from 0.25% to 0.75%, or (ii) a Secured Overnight Financing Rate (“SOFR”) based rate plus a margin ranging from 1.25% to 1.75%. In addition, an unused fee of 0.25% per annum is payable monthly in arrears based on the undrawn portion of the commitments in respect of the 5/3 Revolver. Borrowings under the 5/3 Revolver are secured by a first priority lien in substantially all of the present and future property and assets, real and personal, of the Company, subject to customary exceptions.
Under the 5/3 Revolver, we are subject to certain customary affirmative and negative covenants. In addition, the 5/3 Revolver contains certain financial covenants requiring the Company to maintain, on a consolidated basis as of the last day of each month, a fixed charge coverage ratio of at least 1.10 to 1.0.
Harrow Access for All
In September 2025, we announced Harrow Access For All (“HAFA”) to expand our proprietary patient access model from a single product to encompass Harrow’s comprehensive ophthalmic portfolio of branded, authorized generics (AGx), and compounded ophthalmic medications. Beginning in late 2025 and expanding into 2027, HAFA will provide a single, unified access point for prescribers and patients, offering affordability, streamlined prescribing, and predictable access. The platform creates a simpler, more predictable path to treatment—supporting better outcomes for patients and greater efficiency for physicians.
8.625% Senior Notes Due 2030 and Payoff of Prior Debt
In September 2025, we closed a private offering of $250,000,000, aggregate principal amount of 8.625% senior notes due 2030. The 2030 Notes offering resulted in net proceeds to us of approximately $242,748,000 after deducting underwriting discounts and commissions and other offering expenses of $7,252,000.
The 2030 Notes are senior unsecured obligations and are effectively subordinated to any of our secured indebtedness to the extent of the value of the assets securing such indebtedness. The 2030 Notes are guaranteed on a senior unsecured basis by us, subject to certain exceptions. The 2030 Notes bear interest at the rate of 8.625% per annum. Interest on the 2030 Notes is payable semi-annually in arrears on March 15 and September 15 of each year. The issuance costs were recorded as a debt discount and are being amortized as interest expense over the term of the 2030 Notes using the effective interest rate method.
We used the net proceeds from the 2030 Notes offering to prepay all then outstanding senior debt borrowings, exit costs, and accrued interest including $107,500,000 in total principal loan amount borrowed under the Credit Agreement and Guaranty (the “Oaktree Loan”) with Oaktree Fund Administration, LLC, as administrative agent for the lenders (together, “Oaktree”), $75,000,000 in total principal amount senior notes due 2026 (the “2026 Notes”), and $40,250,000 in total principal amount senior notes due 2027 (the “2027 Notes”). The 2026 Notes and 2027 Notes were listed on The Nasdaq Stock Market under the symbols “HROWL” and “HROWM”, respectively. The 2026 Notes were delisted on October 10, 2025 and the 2027 Notes were delisted on October 8, 2025.
| 53 |
BYOOVIZ® and OPUVIZTM – Commercialization Agreement
In July 2025, we entered into a development and commercialization agreement (the “Samsung Agreement”) with Samsung Bioepis Co., Ltd. (“Samsung”). Under the terms of the Samsung Agreement, following completion of the transition of commercial rights from Biogen, Inc. back to Samsung, Samsung will develop, manufacture, and supply BYOOVIZ (ranibizumab-nuna) and OPUVIZ (aflibercept-yszy) (individually, a “Product” and together, the “Products”) for Harrow to commercialize in the U.S. market (the “Rights”). In consideration of the Rights, we made a one-time upfront payment to Samsung of $4,000,000 in February 2026, and Samsung will be eligible to receive additional one-time payments based on the achievement of net sales-based milestones of the Products. In addition to other mutually agreed terms, we shall pay to Samsung a share of net sales from the Products generated in the U.S. market. We expect BYOOVIZ to be available in the middle of 2026 and OPUVIZ to be available in the middle of 2027.
Acquisition of Commercial Rights to BYQLOVITM
In June 2025, we announced a licensing agreement whereby we acquired the exclusive U.S. commercial rights to BYQLOVI (clobetasol propionate ophthalmic suspension) 0.05% from Taiwan-based Formosa Pharmaceuticals. BYQLOVI was recently approved by the FDA for the treatment of post-operative inflammation and pain following ocular surgery and is the first new ophthalmic steroid in its class in over 15 years. Harrow expects BYQLOVI to be available to launch in the US in the middle of 2026.
VEVYE Access for All
In March 2025, we announced a patient access program called VEVYE Access for All. The program is designed to increase patient access to VEVYE at an out-of-pocket cost of $59 or below and, in many cases, reduce the need for prior authorizations, step edits, and other treatment obstacles facing dry eye patients and their prescribers.
Project Beagle
In March 2025, we initiated a 360-degree review of opportunities to offer ImprimisRx customers a Harrow-owned FDA-approved product alternative to a compounded formulation. We call this initiative Project Beagle. In that vein, we began implementing a continuity of care program to transition approximately 25,000 ImprimisRx patients from our Klarity-C (0.1% cyclosporine) compounded formulation to VEVYE (0.1% cyclosporine), and we discontinued compounding Klarity-C during 2025. We are also discontinued another related compounded formulation called Klarity PF. Klarity PF is primarily purchased by a concentrated group of customers who we expect to continue accepting our FRESHKOTE product as an alternative. In February 2026, we announced the launch of PharmaPack™, a direct-to-prescriber cash-pay offering designed to expand access to affordable, FDA-approved branded ophthalmic therapies as alternatives to compounded formulations. As we work through Project Beagle, we will continue to review opportunities to reduce the size of our compounded formulary, improve and simplify our compounding capabilities, and transition other ImprimisRx customers from compounded formulations to Harrow’s FDA-approved products.
Results of Operations
The following period-to-period comparisons of our financial results are not necessarily indicative of results for any future period.
Comparison of Years Ended December 31, 2025 and 2024
Revenues
Our revenues include amounts recorded from sales of branded products to wholesalers through a third-party logistics facility, sales of proprietary compounded formulations, and revenues received from royalty payments owed to us pursuant to out-license and like arrangements. The following table presents our revenues for the years ended December 31, 2025 and 2024:
| For the Years Ended | ||||||||||||
| December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| IHEEZO net sales | $ | 81,348,000 | $ | 49,303,000 | $ | 32,045,000 | ||||||
| VEVYE net sales | 88,688,000 | 28,061,000 | 60,627,000 | |||||||||
| Other branded products net sales | 25,326,000 | 37,836,000 | (12,510,000 | ) | ||||||||
| Other revenues, net | 394,000 | 915,000 | (521,000 | ) | ||||||||
| Branded revenue, net | 195,756,000 | 116,115,000 | 79,641,000 | |||||||||
| ImprimisRx revenue, net | 76,547,000 | 83,499,000 | (6,952,000 | ) | ||||||||
| Total revenues, net | $ | 272,303,000 | $ | 199,614,000 | $ | 72,689,000 | ||||||
| 54 |
The increase in Branded revenues from product sales between the years ended December 31, 2025 and 2024 was primarily related to an increase in sales and units sold of IHEEZO and VEVYE resulting from increased marketing efforts. These increases were partially offset by lower sales of other brands and lower Imprimis revenue.
The decrease in ImprimisRx revenue was primarily due to a decrease in volume for the year ended December 31, 2025 compared to 2024.
Cost of Sales
Our cost of sales includes direct and indirect costs to manufacture formulations and sell products, including API, personnel costs, packaging, storage, royalties, shipping and handling costs, manufacturing equipment and tenant improvements depreciation, the write-off of obsolete inventory, amortization of acquired product NDAs, and other related expenses.
The following table presents our cost of sales for the years ended December 31, 2025 and 2024:
Branded
| For the Years Ended December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| Cost of sales | $ | 37,230,000 | $ | 21,667,000 | $ | 15,563,000 | ||||||
The increase in Branded cost of sales was primarily attributable to an increase in units sold of IHEEZO and VEVYE during the years ended December 31, 2025 and 2024 as well as an increase in intangible asset amortization related to acquired product rights for TRIESENCE and royalties related to VEVYE and IHEEZO.
ImprimisRx
| For the Years Ended December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| Cost of sales | $ | 30,704,000 | $ | 27,578,000 | $ | 3,126,000 | ||||||
The increase in ImprimisRx costs of sales between the years ended December 31, 2025 and 2024 was primarily attributable to product mix that included more sales of lower gross margin products and inventory losses.
Gross Profit and Margin
Branded
| For the Years Ended December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| Gross profit | $ | 158,526,000 | $ | 94,448,000 | $ | 64,078,000 | ||||||
| Gross margin | 81.0 | % | 81.3 | % | (0.3 | )% | ||||||
| 55 |
Gross Margin increased due to increased sales. The slight decrease in Branded gross margin percentage between the years ended December 31, 2025 and 2024 was primarily attributable to an increase in our fixed expenses, in particular, acquired product rights amortization related to the launch of TRIESENCE and a related contingent milestone payment that was capitalized in the fourth quarter of 2024.
ImprimisRx
| For the Years Ended December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| Gross profit | $ | 45,843,000 | $ | 55,921,000 | $ | (10,078,000 | ) | |||||
| Gross margin | 59.9 | % | 67.0 | % | (7.1 | )% | ||||||
ImprimisRx gross margin decreased during the year ended December 31, 2025 compared to 2024 due to the previously mentioned change in product mix as well as inventory losses from lower manufacturing efficiency.
Selling, General and Administrative Expenses
Our selling, general and administrative (“SG&A”) expenses include personnel costs, including wages and stock-based compensation, corporate facility expenses, and investor relations, consulting, insurance, filing, legal and accounting fees and expenses as well as costs associated with our marketing activities and sales of our proprietary compounded formulations and other non-proprietary pharmacy products and formulations.
The following table presents our SG&A expenses for the years ended December 31, 2025 and 2024:
| For the Years Ended December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| Selling, general and administrative | $ | 152,914,000 | $ | 129,064,000 | $ | 23,850,000 | ||||||
The increase in SG&A expenses between the years ended December 31, 2025 and 2024 was primarily attributable to (1) increased payroll and related expenses of $15,092,000 due to the addition of new employees in sales, marketing and other departments to support current and expected growth, (2) increased marketing and advertising expense of $3,600,000 and (3) increased audit fees as a result of the Company being subject to the audit attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. These increases were partially offset by a decrease in stock-based compensation expense of $5,114,000 between the periods.
Research and Development Expenses
Our R&D expenses primarily included personnel costs, including wages and stock-based compensation, expenses related to the development of intellectual property, investigator-initiated research and evaluations, formulation development, acquired in-process R&D and other costs related to the clinical development of our assets.
The following table presents our R&D expenses for the years ended December 31, 2025 and 2024:
| For the Years Ended December 31, | $ | |||||||||||
| 2025 | 2024 | Variance | ||||||||||
| Research and development | $ | 20,940 ,000 | $ | 12,230,000 | $ | 8,710,000 | ||||||
The increase in R&D expenses between the years ended December 31, 2025 and 2024 was primarily attributable to one time in-process R&D expense related to the acquisition of Melt of $8,450,000 during the fourth quarter of 2025. In addition, increased development activity related to our branded product portfolio, new product candidate development efforts, and clinical and medical support. During the fourth quarter of 2024, we recorded $2,000,000 of one-time R&D costs associated with the product development of TRIESENCE.
| 56 |
Impairment and Disposal of Long-Lived Assets
During the year ended December 31, 2025, there were no impairments or disposals of long-lived assets. During the year ended December 31, 2024, we recognized an impairment loss of $253,000 related to intellectual property that we expect to no longer utilize in future revenue generating products and compounded formulations.
Interest Expense, net
Interest expense, net was $24,180,000 during the year ended December 31, 2025, compared to $22,786,000 during the year ended December 31, 2024. The increase was primarily due to an increase in the principal balance of our loans over the periods presented.
Investment Gain (Loss) from Eton
During the year ended December 31, 2025, there was no gain (loss) from investments. During the year ended December 31, 2024, we recorded a loss of $3,171,000 related to the change in fair market value of Eton’s common stock at the time of its sale, including trading expenses and commissions of approximately $436,000. In April 2024, we sold all of our remaining shares in Eton.
Loss on Early Extinguishment of Debt
During the year ended December 31, 2025, we recorded a loss on extinguishment of debt of $7,750,000 related to the payoff of a loan. There were no extinguishments of debt during the year ended December 31, 2024.
Other Income (Expense), net
During the year ended December 31, 2025, other income of $47,000 represents foreign exchange gains on settlement of foreign-denominated payables. During the year ended December 31, 2024, we recorded other expense, net, of $185,000 related primarily to income from the sublease of office space in Nashville, offset by a loss associated with a cybersecurity incident.
Tax Expense
During the years ended December 31, 2025 and 2024, we recorded income tax expense of $3,771,000 and $161,000, respectively. The increase in income tax expense in 2025 was primarily related to limitations of stock-based compensation expense under Section 179(m) of the Internal Revenue Code.
The following table presents our net loss for the years ended December 31, 2025 and 2024:
| For the Years Ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Net loss | $ | (5,139,000 | ) | $ | (17,481,000 | ) | ||
| Net loss per share, basic and diluted | $ | (0.14 | ) | $ | (0.49 | ) | ||
Liquidity and Capital Resources
Liquidity
Our cash on hand at December 31, 2025 was $72,927,000, compared to $47,247,000 at December 31, 2024.
As of the date of this Annual Report, we believe that cash and cash equivalents of $72,927,000 at December 31, 2025 will be sufficient to sustain our planned level of operations and capital expenditures for fiscal year 2026 and the foreseeable future. We may consider the sale of certain assets including, but not limited to, part of, or all of, our investment in Surface and any of our consolidated subsidiaries. However, we may pursue acquisitions of products, drug candidates or other strategic transactions that involve large expenditures or we may experience growth more rapidly or on a larger scale than we expect, any of which could result in the depletion of capital resources more rapidly than anticipated and could require us to seek additional financing to support our operations.
| 57 |
We expect to use our current cash position and funds generated from our operations and any financing to pursue our business plan, which includes developing and commercializing products, drug candidates, compounded formulations and technologies, integrating and developing our operations, pursuing potential future strategic transactions as opportunities arise, including potential acquisitions of additional drug products, drug candidates, and/or assets or technologies, pharmacies, outsourcing facilities, drug company and manufacturers, and otherwise fund our operations. We may also use our resources to conduct clinical trials or other studies in support of our formulations or any drug candidate for which we pursue FDA approval, to pursue additional development programs or to explore other development opportunities.
Net Cash Flows
The following provides detailed information about our net cash flows for the years ended December 31, 2025, 2024 and 2023:
| For the Years Ended December 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Net cash provided by (used in): | ||||||||||||
| Operating activities | $ | 43,864,000 | $ | (22,202,000 | ) | $ | 3,840,000 | |||||
| Investing activities | (5,460,000 | ) | (33,164,000 | ) | (152,553,000 | ) | ||||||
| Financing activities | (12,724,000 | ) | 28,528,000 | 126,528,000 | ||||||||
| Net change in cash and cash equivalents | 25,680,000 | (26,838,000 | ) | (22,185,000 | ) | |||||||
| Cash and cash equivalents at beginning of the period | 47,247,000 | 74,085,000 | 96,270,000 | |||||||||
| Cash and cash equivalents at end of the year | $ | 72,927,000 | $ | 47,247,000 | $ | 74,085,000 | ||||||
Operating Activities
Net cash provided by operating activities was $43,864,000 in 2025, compared to cash used in of $22,202,000 in the prior year. The increase in net cash provided by operating activities between the periods was mainly attributed to better operating results and collections of $5,010,000 of accounts receivable as a result of collection efforts partially offset by settlement of accrued accounts payable invoices of $4,608,000.
Net cash used in operating activities was $22,202,000 in 2024, compared to cash provided by of $3,840,000 in the prior year. The decrease in net cash provided by operating activities between the periods was mainly attributed to changes in our working capital balances including accounts payable, prepaid expenses, inventories and most notably, accounts receivable. Our accounts receivable balance between periods increased significantly due to an increase in our branded product sales, which have a longer revenue cycle compared to our ImprimisRx product sales. In addition, during 2024, we extended additional terms to our largest distributor to allow for downstream and end users (e.g. hospitals, clinics and ambulatory surgery centers) of certain of our branded products additional time to pay for our branded products.
Investing Activities
Net cash used in investing activities in 2025 and 2024 was $5,460,000 and $33,164,000, respectively. Cash used in investing activities in 2025 was primarily due the acquisition of Melt for $4,358,000 and equipment and software purchases of $887,000. Cash used in investing activities in 2024 was primarily due to the milestone payment of $37,000,000 related to TRIESENCE partially offset by cash received from the sale of our investment in Eton for $5,510,000. Cash used in investing activities in 2023 was primarily associated with product acquisitions.
| 58 |
Financing Activities
Net cash used in financing activities in 2025 was $12,724,000 and cash provided by financing activities in 2024 was $28,528,000. Cash used in financing activities during the year ended December 31, 2025 was primarily due to repayment of debt and payment of payroll taxes upon vesting of stock compensation mostly offset by proceeds from issuance of new debt. Cash provided by financing activities during the year ended December 31, 2024 was primarily due to additional borrowings under our long-term debt facility with Oaktree of $29,780,000, net of issuance costs, and proceeds from the exercise of stock options, offset by the payment of taxes associated with the vesting and exercise of share-based awards. Cash provided by financing activities during the year ended December 31, 2023 was primarily related to proceeds received from the issuance of the Oaktree Loan and Oaktree Amendment, issuance of unsecured debt and sale of our equity, offset by payment of payroll taxes upon vesting of PSUs in exchange for shares withheld from employees.
Sources of Capital
During the year ended December 31, 2025, our principal sources of cash came from cash generated by our operating activities. In future periods, including the year ending December 31, 2026, we expect cash to be provided from our operating activities, but our forecasts may not be accurate and our plans may change. We may also sell some or all of our ownership interests in Surface or our other subsidiaries
In September 2025, we completed the sale of the 2030 Notes in a private offering and received net proceeds of $242,748,000. We used the net proceeds from the 2030 Notes to prepay all outstanding borrowings under the Oaktree Loan, the 2027 Notes, and the 2026 Notes, and to pay certain exit costs related thereto. The remaining funds will be used for general corporate purposes, which may include funding future strategic business development opportunities and related investments. We also entered into the 5/3 Revolver with Fifth Third in September 2025, which provided the Company with a secured revolving credit facility of $40,000,000, with an additional $20,000,000 of uncommitted incremental revolving line of credit. We have not drawn down any amounts under the 5/3 Revolver. The Company is in compliance with all debt covenants and expects to remain compliant for at least the next 12 months.
We may acquire new products, product candidates and/or businesses and, as a result, we may need significant additional capital to support our business plan and fund our proposed business operations. We may receive additional proceeds from the exercise of stock purchase warrants that are currently outstanding. We may also seek additional financing from a variety of sources, including other equity or debt financings, funding from corporate partnerships or licensing arrangements, sales of assets or any other financing transaction. If we issue equity or convertible debt securities to raise additional funds, our existing stockholders may experience substantial dilution, and the newly issued equity or debt securities may have more favorable terms or rights, preferences and privileges senior to those of our existing stockholders. If we raise additional funds through collaboration or licensing arrangements or sales of assets, we may be required to relinquish potentially valuable rights to our product candidates or proprietary technologies or formulations, or grant licenses on terms that are not favorable to us. If we raise funds by incurring additional debt, we may be required to pay significant interest expenses and our leverage relative to our earnings or to our equity capitalization may increase. Obtaining commercial loans, assuming they would be available, would increase our liabilities and future cash commitments and may impose restrictions on our activities, such as the financial and operating covenants. Further, we may incur substantial costs in pursuing future capital and/or financing transactions, including investment banking fees, legal fees, accounting fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we may issue, such as convertible notes and warrants, which would adversely impact our financial results.
We may be unable to obtain financing when necessary as a result of, among other things, our performance, general economic conditions, conditions in the pharmaceuticals and pharmacy industries, or our operating history. In addition, the fact that we have a limited history of profitability could further impact the availability or cost to us of future financings. As a result, sufficient funds may not be available when needed from any source or, if available, such funds may not be available on terms that are acceptable to us. If we are unable to raise funds to satisfy our capital needs when needed, then we may need to forego pursuit of potentially valuable development or acquisition opportunities, we may not be able to continue to operate our business pursuant to our business plan, which would require us to modify our operations to reduce spending to a sustainable level by, among other things, delaying, scaling back or eliminating some or all of our ongoing or planned investments in corporate infrastructure, business development, sales and marketing and other activities, or we may be forced to discontinue our operations entirely.
| 59 |
Critical Accounting Policies and Estimates
We rely on the use of estimates and make assumptions that impact our financial condition and results. These estimates and assumptions are based on historical results and trends as well as our forecasts of how results and trends might change in the future. Although we believe that the estimates we use are reasonable, actual results could differ materially from these estimates.
We believe that the accounting policies described below are critical to understanding our business, results of operations and financial condition because they involve the use of more significant judgments and estimates in the preparation of our consolidated financial statements. An accounting policy is deemed to be critical if it requires an accounting estimate to be made based on assumptions about matters that are highly uncertain at the time the estimate is made, and any changes in the assumptions used in making the accounting estimates that are reasonably likely to occur could materially impact our consolidated financial statements.
Revenue Recognition
We account for contracts with customers in accordance with ASC 606, Revenues from Contracts with Customers. We have two primary streams of revenue: (1) product revenues, including revenue recognized from sales of products through its pharmacy and outsourcing facility and sales of branded products to wholesalers through a third-party logistics (“3PL”) partner, and (2) revenue recognized from intellectual property licenses.
Product Revenues
We recognize revenue from product sales at a point in time when our customer is deemed to have obtained control of the product, which generally occurs upon receipt or acceptance by our customer.
Sales of branded pharmaceutical products are subject to variable consideration due to chargebacks, government rebates, returns, administrative and other rebates, and cash discounts. Estimates for these elements of variable consideration require significant judgment.
We record reserves for rebates, chargebacks, discounts, distribution fees and product returns at the time revenue is recognized. These reserves are inherently uncertain because they depend on future utilization patterns, payor mix, wholesaler inventory levels, and contractual terms that vary across customers and programs. We use historical experience (where available), current-period data from distributors and payors, and forecasted sales volumes to estimate these amounts.
At December 31, 2025, our sales deduction and returns reserves totaled $68,381,000 compared to $59,631,000 at December 31, 2024. The increase primarily reflects (i) higher ophthalmic product sales, (ii) expanded commercial rebate programs, and (iii) higher expected product returns associated with the timing of lot expirations, partially offset by lower co-pay assistance costs.
Our estimates are sensitive to changes in payor mix and program utilization. For example, if our aggregate rebate and discount rate for 2025 had been 2 percentage points higher, product revenue would have been approximately $10.6 million lower, and if it had been 2 percentage points lower, product revenue would have been approximately $10.6 million higher.
Income Taxes
As part of the process of preparing our consolidated financial statements, we must estimate the actual current tax assets and liabilities and assess permanent and temporary differences that result from differing treatment of items for tax and accounting purposes. The temporary differences result in deferred tax assets and liabilities, which are included within the consolidated balance sheets. A valuation allowance is established for deferred tax assets for which it is more likely than not that some portion or all of the deferred tax assets will not be realized. We periodically re-assess the need for a valuation allowance against our deferred tax assets based on all available evidence, including scheduled reversals of deferred tax liabilities, projected future taxable income, tax planning strategies, results of recent operations, and our historical earnings experience by taxing jurisdiction. Significant judgment is required in making this assessment.
| 60 |
We recognize the financial statement effects of a tax position when our assessment is that there is more than a 50% probability that the position will be sustained upon examination by a taxing authority based upon its technical merits. Uncertain tax positions are recorded based upon certain recognition and measurement criteria. Significant judgment is required in making this assessment, and, therefore, we re-evaluate uncertain tax positions and consider various factors, including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, information obtained during in-process audit activities, and changes in facts or circumstances related to a tax position. We adjust the amount of the liability to reflect any subsequent changes in the relevant facts and circumstances surrounding the uncertain tax positions.
Intangible Assets
Intangible assets acquired in a business combination are recorded at fair value, while intangible assets acquired in connection with an asset acquisition are recorded at cost. Payments to acquire intangible assets in an asset acquisition may include up-front payments and contingent consideration. With regard to contingent consideration in an asset acquisition, the Company recognizes regulatory milestones upon achievement, royalties in the period in which the underlying sales occur, and sales-based milestones when the milestone is deemed probable by the Company of being achieved. Significant judgment is involved in assessing the probability of achievement of milestones. If contingent consideration is recognized subsequent to the acquisition date in an asset acquisition, the amount of such consideration is recorded as an addition to the cost basis of the intangible asset and amortization expense is recorded prospectively over the remaining useful life of the asset.
Impairment of Intangible Assets
We hold significant definite lived intangible assets including; product rights, licensed intangible assets, and other intangible assets. Under GAAP, we evaluate these assets if events or changes in circumstances indicate that the carrying amount may not be recoverable (e.g., lower-than-expected sales, adverse regulatory or competitive developments, higher required returns, or changes in macroeconomic conditions).
When we test definite-lived assets, we compare the carrying value to the undiscounted future cash flows expected to result from the use and eventual disposition of the asset group. If those cash flows are less than the carrying amount, we recognize an impairment equal to the amount by which the carrying value exceeds fair value.
As a result of its assessment in 2024 and 2023, we recorded an impairment charge of $253,000 and $380,000, respectively, related to the impairment of certain licenses, trademarks, patents and patent applications (see Note 11 to our consolidated financial statements). We did not recognize any impairment charges for the year ended December 31, 2025.
Stock-Based Compensation
We measure stock-based compensation for stock options, restricted stock units (“RSUs”), performance stock units (“PSUs”), warrants, and restricted stock at fair value on the grant date and recognize the associated expense over the requisite service period. Estimating the grant-date fair value requires significant judgment because the valuation models we use—including the Black-Scholes-Merton option-pricing model and Monte Carlo simulation models for certain performance awards—depend on several subjective assumptions such as expected volatility, expected term, risk-free interest rates, dividend yield, and, for PSUs, the probability of achieving market-based performance targets.
These assumptions are inherently uncertain because they rely on forward-looking estimates of employee exercise behavior, future share price volatility, and performance outcomes that may differ from actual experience. For example, expected volatility is based on historical volatility of our stock and those of comparable companies, which may not be indicative of future results. Likewise, the estimated forfeiture rate reflects management’s judgment regarding employee turnover and achievement of service or performance conditions and is revised when actual results differ from initial expectations.
| 61 |
Our stock-based compensation expense is sensitive to changes in these inputs. Holding all other assumptions constant, a 10% increase in the expected volatility used to value stock option grants would increase the grant-date fair value of such awards by approximately 7%, and a 10% decrease in the expected term would decrease the fair value by approximately 7%. Actual outcomes that differ from our assumptions, including changes in share price performance or employee turnover, could materially affect the amount and timing of stock-based compensation expense in future periods.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, including the use of structured finance, special purpose entities or variable interest entities.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Market risk is the potential loss arising from adverse changes in market rates and prices, such as interest rates. Our exposure to market risk is limited and relates primarily to interest rate risk on our cash and cash equivalents and the fair value of our outstanding fixed-rate indebtedness
Interest Rate Risk
As of December 31, 2025, all of our outstanding indebtedness bears interest at fixed rates. Accordingly, changes in market interest rates do not affect our contractual cash interest obligations or debt service requirements. However, changes in interest rates may affect the fair value of our fixed-rate debt. Based on our outstanding fixed-rate indebtedness as of December 31, 2025, a hypothetical 100 basis point movement in market interest rates would change the estimated fair value of such debt by approximately $2.5 million. These estimated changes would not impact our consolidated statements of operations or cash flows unless the debt is refinanced, repurchased, or otherwise settled prior to the maturity.
Our cash and cash equivalents consist primarily of demand deposits and other highly liquid instruments with short-term maturities. As a result, interest income earned on these balances may fluctuate with changes in short-term interest rates. We do not believe that reasonably likely changes in interest rates would have a material effect on our consolidated financial position, results of operations, or cash flows.
We do not use derivative financial instruments, including interest rate swaps, to manage interest rate risk.
Foreign Currency and Other Market Risks
We do not have material exposure to foreign currency exchange rate risk, commodity price risk, or other market risks.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements and supplementary data required by this item are included in this Annual Report beginning on page F-1 immediately following the signature page hereto and are incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, under the supervision and with the participation of our CEO, our principal executive officer, and our CFO, our principal financial officer, conducted an evaluation of the effectiveness of our disclosure controls and procedures as of December 31, 2025, the end of the period covered by this Annual Report, pursuant to Rules 13a-15(b) and 15d-15(b) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”).
| 62 |
In connection with that evaluation, our CEO and CFO concluded that, as of December 31, 2025, our disclosure controls and procedures were effective. For the purpose of this review, disclosure controls and procedures mean controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. These disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to management, including our principal executive officer, principal financial officer and principal accounting officer, as appropriate to allow timely decisions regarding required disclosure.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Internal control over financial reporting is a process designed by, or under the supervision of, our CEO and CFO and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our management, under the supervision and with the participation of our CEO and CFO, conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations. Based on such evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2025.
Deloitte & Touche LLP, the independent registered public accounting firm who also audited our Consolidated Financial Statements for 2025, has issued an attestation report on the Company’s effectiveness of internal controls over financial reporting which is included herein. The report by Deloitte & Touche LLP is included in our consolidated financial statements beginning on page F-1 of this report.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) during the three months ended December 31, 2025, that has materially affected, or is reasonably likely to materially affect our internal control over financial reporting.
Inherent Limitations on Effectiveness of Controls
Our management, including our CEO and CFO, do not expect that our disclosure controls or our internal control over financial reporting will prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. The design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Further, because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Projections of any evaluation of controls effectiveness to future periods are subject to risks. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures.
ITEM 9B. OTHER INFORMATION
From
time to time, certain of our executive officers and directors may enter into, amend or terminate written trading arrangements pursuant
to Rule 10b5-1 of the Exchange Act or otherwise. During the three months ended December 31, 2025, none of our directors or officers
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
| 63 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated by reference to the information set forth under the captions “Election of Directors,” “Executive Officers,” “Corporate Governance,” “Corporate Governance — Delinquent Section 16(a) Reports,” and “Corporate Governance — Code of Business Conduct and Ethics” in the Company’s Proxy Statement for the 2026 Annual Meeting of Stockholders.
We have adopted an Insider Trading Policy governing transactions in our securities by all officers of the Company and its subsidiaries, all members of the Company’s Board of Directors and all employees of the Company and its subsidiaries, and we believe such policy is reasonably designed to promote compliance with insider trading laws, rules and regulations, and the exchange listing standards applicable to us. A copy of our Insider Trading Policy is filed as Exhibit 19 to this Annual Report on Form 10-K. It is our policy to comply with all applicable securities laws and regulations (including appropriate approvals by our Board of Directors, if required) when engaging in transactions in our securities.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated by reference to the information in the Company’s Proxy Statement for the 2026 Annual Meeting of Stockholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference to the information set forth under the captions “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” and “Executive Compensation — Securities Authorized for Issuance Under Equity Compensation Plans” in the Company’s Proxy Statement for the 2026 Annual Meeting of Stockholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated by reference to the information set forth under the captions “Corporate Governance — Transactions with Related Persons” and “Corporate Governance — Director Independence” in the Company’s Proxy Statement for the 2026 Annual Meeting of Stockholders.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated by reference to the information set forth under the caption “Ratification of Selection of Independent Registered Public Accounting Firm” in the Company’s Proxy Statement for the 2026 Annual Meeting of Stockholders.
| 64 |
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
(a) |
List of the following documents filed as part of the report: |
|
(1) |
See the index to our consolidated financial statements on page F-1 for a list of the financial statements being filed in this Annual Report. |
|
|
|
|
(2) |
All financial statement schedules are omitted because they are not applicable or the required information is shown in the consolidated financial statements or the notes thereto. |
|
|
|
|
(3) |
See Item 15(b) below for all exhibits being filed or incorporated by reference herein. |
|
(b) |
Exhibits: |
EXHIBIT INDEX
Exhibit No. |
|
Description |
2.1 |
||
3.1 |
|
|
3.2 |
|
|
4.1* |
|
|
4.2 |
|
|
4.3 |
|
|
4.4 |
Form of 8.625% Senior Note due 2026 (included as Exhibit A in Exhibit 4.3). | |
4.5 |
|
|
4.6 |
|
Form of 11.875% Senior Note due 2027 (included as Exhibit A in Exhibit 4.5). |
4.7 |
|
|
4.8 |
|
Form of 8.625% Senior Note due 2030 (included in Exhibit 4.7). |
10.1 |
||
10.2# |
|
|
10.3# |
|
| 65 |
10.4# |
|
|
10.5# |
|
|
10.6# |
|
|
10.7# |
|
|
10.8# |
|
|
10.9# |
|
|
10.10# |
|
|
10.11# |
|
|
10.12# |
|
|
10.13# |
|
|
10.14# |
|
|
10.15#* |
|
|
10.16 |
|
|
10.17# |
|
|
10.18# |
|
|
10.19# |
|
|
10.20 |
|
|
10.21*+ |
|
|
10.22 |
|
| 66 |
10.23 |
|
|
10.24 |
|
|
10.25 |
|
|
10.26 |
|
|
10.27 |
|
|
10.28*+ |
|
|
10.29*+ |
|
|
| 10.30#* | Consulting Agreement dated March 1, 2026 between Harrow, Inc. and John P. Saharek | |
16.1 |
|
|
16.2 |
|
|
19 |
|
|
21.1* |
|
|
23.1* |
|
Consent of Independent Registered Public Accounting Firm - Deloitte & Touche LLP |
23.2* |
|
Consent of Independent Registered Public Accounting Firm - Crowe LLP |
23.3* |
|
Consent of Independent Registered Public Accounting Firm – KMJ Corbin & Company LLP |
24.1* |
|
Power of Attorney (included on the signature page to this Annual Report) |
31.1* |
|
|
31.2* |
|
|
32.1** |
|
|
32.2** |
|
|
97 |
|
|
101* |
|
The following financial information from the Company’s Annual Report on Form 10-K for the year ended December 31, 2025, formatted in Inline Extensible Business Reporting Language (iXBRL): (i) the Balance Sheets, (ii) the Statements of Operations, (iii) the Statement of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit), (iv) the Statements of Cash Flows and (v) Notes to Financial Statements. |
104* |
|
The cover page from the Company’s Annual Report on Form 10-K for the year ended December 31, 2025 has been formatted in Inline XBRL (included as Exhibit 101) |
# |
Management contract or compensatory plan or arrangement. |
* |
Filed herewith. |
** |
Furnished herewith. |
+ |
Certain portions of this exhibit have been omitted pursuant to Item 601(a)(5) and/or the redacted exhibit rules under Item 601(b)(2) and/or Item 601(b)(10)(iv) of Regulation S-K because they are not material and are the type of information that the registrant customarily and actually treats as private or confidential / and would likely cause competitive harm if publicly disclosed. The registrant agrees to furnish supplementally an unredacted copy of any exhibit marked with “+” to the SEC upon request. |
|
Omitted schedules/attachments: Schedules and similar attachments to exhibits have been omitted pursuant to Item 601(a)(5). The registrant hereby undertakes to furnish copies of any omitted schedules or attachments to the SEC upon request. |
ITEM 16. FORM 10-K SUMMARY
None.
| 67 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
HARROW, INC. | |
|
|
|
|
By: |
/s/ Mark L. Baum |
|
|
Mark L. Baum |
|
|
Chief Executive Officer (Principal Executive Officer) |
|
|
|
|
Date: |
March 2, 2026 |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Mark L. Baum and Andrew R. Boll, and each of them individually, as his true and lawful attorneys-in-fact and agents with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities to any or all amendments to this Annual Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents or any of them the full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the foregoing, as full to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or any of them, or his substitutes, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature |
|
Title |
|
Date |
|
|
|
|
|
/s/ Mark L. Baum |
|
Chief Executive Officer and Chairman of the Board |
|
March 2, 2026 |
Mark L. Baum |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ Andrew R. Boll |
|
President and Chief Financial Officer |
|
March 2, 2026 |
Andrew R. Boll |
|
(Principal Financial Officer) |
|
|
|
|
|
|
|
/s/ Randall E. Pollard |
Chief Accounting Officer |
March 2, 2026 | ||
Randall E. Pollard |
|
(Principal Accounting Officer) |
|
|
|
|
|
|
|
/s/ Adrienne L. Graves |
|
Director |
|
March 2, 2026 |
Adrienne L. Graves |
|
|
|
|
|
|
|
|
|
/s/ Lauren P. Silvernail |
|
Director |
|
March 2, 2026 |
Lauren P. Silvernail |
|
|
|
|
|
|
|
|
|
/s/ Perry J. Sternberg |
|
Director |
|
March 2, 2026 |
Perry J. Sternberg |
|
|
|
|
| 68 |
FINANCIAL STATEMENTS
Harrow, Inc.
Index to Consolidated Financial Statements
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the stockholders and the Board of Directors of Harrow, Inc
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of Harrow, Inc (the “Company”) as of December 31, 2025, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2025, based on criteria established in Internal Control — Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated financial statements as of and for the year ended December 31, 2025, of the Company and our report dated March 2, 2026, expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/
March 2, 2026
| F-2 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the stockholders and the Board of Directors of Harrow, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Harrow, Inc. (the “Company”) as of December 31, 2025, the related consolidated statements of operations, stockholders’ equity, and cash flows, for the period ended December 31, 2025, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2025, and the results of its operations and its cash flows for the period ended December 31, 2025, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2025, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 2, 2026, expressed an unqualified opinion on the Company’s internal control over financial reporting.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current-period audit of the financial statements that was communicated or required to be communicated to the audit committee and that (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
| F-3 |
Revenues – Estimates of Government Rebate Accruals – Refer to Note 3 to the financial statements
Critical Audit Matter Description
The Company’s revenues for branded pharmaceutical products are subject to variable consideration, including government rebates. Reserves for rebates are estimated at the time revenue is recognized and are recorded as a reduction to gross revenues and an increase to accrued rebates to governmental agencies for utilization of the Company’s products by beneficiaries under such governmental programs. Estimating these reserves requires significant management judgment because they depend on assumptions such as future utilization patterns, payor mix, wholesaler inventory levels, and contractual terms that vary across customers and programs. The Company uses historical experience (where available), current-period data from distributors and payors, and forecasted sales volumes to estimate these amounts.
We identified the estimates for government rebate reserves as a critical audit matter due to the judgments required and complexity involved in determining the significant assumptions used in calculating the accruals. Auditing these estimates involved a high degree of auditor judgment and an increased extent of effort.
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to government rebate accruals included the following, among others:
| ● | We tested the effectiveness of internal controls over government rebate accruals, including management’s controls over the underlying assumptions used to estimate government rebate reserves. |
| ● | We evaluated the Company’s methods and assumptions used to calculate government rebate accruals, including assumptions of utilization patterns, payor mix, wholesaler inventory levels, and contractual terms. |
| ● | We evaluated the Company’s ability to estimate government rebate accruals accurately by comparing actual amounts incurred for government rebate accruals to historical estimates. |
| ● | We tested the reasonableness of the government rebate accruals recorded at period end by developing an independent expectation for comparison to actual recorded balances. |
/s/
March 2, 2026
We have served as the Company’s auditor since 2025.
| F-4 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Stockholders and the Board of Directors of Harrow, Inc.
Nashville, Tennessee
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Harrow, Inc. (the “Company”) as of December 31, 2024, the related consolidated statements of operations, stockholders’ equity, and cash flows for the year then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2024, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
| /s/ |
We served as the Company’s auditor from 2024 to 2025.
March 27, 2025
| F-5 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Harrow, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of operations, stockholders’ equity, and cash flows of Harrow, Inc. (the “Company”) for the year ended December 31, 2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the results of operations and cash flows of the Company for the year ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/
We served as the Company’s auditor from 2007 to 2024.
March 19, 2024 (March 27, 2025 as to Note 19)
| F-6 |
HARROW, INC.
CONSOLIDATED BALANCE SHEETS
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Accounts receivable, net | ||||||||
| Inventories | ||||||||
| Prepaid expenses and other current assets | ||||||||
| Total current assets | ||||||||
| Property, plant and equipment, net | ||||||||
| Capitalized software costs, net | ||||||||
| Operating lease right-of-use assets, net | ||||||||
| Intangible assets, net | ||||||||
| Goodwill | ||||||||
| TOTAL ASSETS | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Accrued rebates and copay assistance | ||||||||
| Accrued payroll and related liabilities | ||||||||
| Deferred revenue and customer deposits | ||||||||
| Current portion of operating lease obligations | ||||||||
| Total current liabilities | ||||||||
| Operating lease obligations, net of current portion | ||||||||
| Notes payable, net of unamortized debt discount | ||||||||
| TOTAL LIABILITIES | ||||||||
| Commitments and contingencies | ||||||||
| STOCKHOLDERS’ EQUITY | ||||||||
| Common stock, $ par value, shares authorized; and shares issued and outstanding at December 31, 2025 and December 31, 2024, respectively | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| TOTAL HARROW, INC. STOCKHOLDERS’ EQUITY | ||||||||
| Noncontrolling interests | ( | ) | ( | ) | ||||
| TOTAL STOCKHOLDERS’ EQUITY | ||||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-7 |
HARROW, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| For the Years Ended December 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Revenues: | ||||||||||||
| Product sales, net | $ | $ | $ | |||||||||
| Other revenues | ||||||||||||
| Total revenues | ||||||||||||
| Cost of sales | ( | ) | ( | ) | ( | ) | ||||||
| Gross profit | ||||||||||||
| Operating expenses: | ||||||||||||
| Selling, general and administrative | ||||||||||||
| Research and development | ||||||||||||
| Impairment of long-lived assets | ||||||||||||
| Total operating expenses | ||||||||||||
| Income from operations | ||||||||||||
| Other (expense) income: | ||||||||||||
| Interest expense, net | ( | ) | ( | ) | ( | ) | ||||||
| Investment (loss) gain from Eton Pharmaceuticals | ( | ) | ||||||||||
| Loss on extinguishment of debt | ( | ) | ( | ) | ||||||||
| Other income (expense), net | ( | ) | ( | ) | ||||||||
| Total other expense, net | ( | ) | ( | ) | ( | ) | ||||||
| Loss before income taxes | ( | ) | ( | ) | ( | ) | ||||||
| Income tax expense | ( | ) | ( | ) | ( | ) | ||||||
| Net loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Basic and diluted net loss per share of common stock | $ | ) | $ | ) | $ | ) | ||||||
| Weighted-average number of common stock outstanding, basic and diluted | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-8 |
HARROW, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
For the Years Ended December 31, 2025, 2024 and 2023
| Total | Total | |||||||||||||||||||||||||||
| Common Stock | Additional | Harrow, Inc. | Noncontrolling | Total | ||||||||||||||||||||||||
| Par | Paid-in | Accumulated | Stockholders’ | Interest | Stockholders’ | |||||||||||||||||||||||
| Shares | Value | Capital | Deficit | Equity | Equity | Equity | ||||||||||||||||||||||
| Balance at January 1, 2023 | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | ||||||||||||||||||
| Issuance of common stock in connection with: | ||||||||||||||||||||||||||||
| Public offering, net of offering costs | ||||||||||||||||||||||||||||
| Exercise of consultant stock-based options | ||||||||||||||||||||||||||||
| Exercise of employee stock-based options | ||||||||||||||||||||||||||||
| Vesting of RSUs and PSUs | ( | ) | ||||||||||||||||||||||||||
| Shares withheld related to net share settlement of equity awards | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||
| Stock-based compensation expense | - | |||||||||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | ||||||||||||||||||
| Issuance of common stock in connection with: | ||||||||||||||||||||||||||||
| Exercise of employee stock-based options | ||||||||||||||||||||||||||||
| Vesting of RSUs and PSUs | ||||||||||||||||||||||||||||
| Shares withheld related to net share settlement of equity awards | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||
| Stock-based compensation expense | - | |||||||||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | ||||||||||||||||||
| Issuance of common stock in connection with: | ||||||||||||||||||||||||||||
| Exercise of employee stock-based options | ||||||||||||||||||||||||||||
| Vesting of PSUs and RSUs | ( | ) | ||||||||||||||||||||||||||
| Shares withheld related to net share settlement of equity awards | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||
| Stock-based compensation expense | - | |||||||||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||
| Balance at December 31, 2025 | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | ||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-9 |
HARROW, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| For the Years Ended December 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||||||
| Net loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | ||||||||||||
| Depreciation and amortization of property, plant and equipment and software development costs | ||||||||||||
| Amortization of intangible assets | ||||||||||||
| Non-cash lease expense | ||||||||||||
| Acquisition of Melt Pharmaceuticals | ||||||||||||
| Provision for credit losses | ||||||||||||
| Amortization of debt issuance costs and debt discount | ||||||||||||
| Investment loss (gain) from investment in Eton | ( | ) | ||||||||||
| Loss on disposal of equipment | ||||||||||||
| Impairment of intangible assets | ||||||||||||
| Loss on extinguishment of debt | ||||||||||||
| Stock-based compensation | ||||||||||||
| Changes in assets and liabilities: | ||||||||||||
| Accounts receivable | ( | ) | ( | ) | ||||||||
| Inventories | ( | ) | ( | ) | ||||||||
| Prepaid expenses and other current assets | ( | ) | ( | ) | ||||||||
| Accounts payable, accrued expenses, accrued rebates and copay assistance | ( | ) | ||||||||||
| Accrued payroll and related liabilities | ||||||||||||
| Deferred revenue and customer deposits | ( | ) | ( | ) | ||||||||
| NET CASH PROVIDED BY (USED IN) OPERATING ACTIVITIES | ( | ) | ||||||||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||||||
| Net proceeds on sale of investment in Eton Pharmaceuticals | ||||||||||||
| Acquisition of Melt Pharmaceuticals | ( | ) | ||||||||||
| Investment in patent and trademark assets | ( | ) | ( | ) | ||||||||
| Purchase of product rights and related patents | ( | ) | ( | ) | ( | ) | ||||||
| Purchases of property, plant and equipment | ( | ) | ( | ) | ( | ) | ||||||
| NET CASH USED IN INVESTING ACTIVITIES | ( | ) | ( | ) | ( | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||||||
| Net proceeds from 11.875% notes payable, net of costs | ||||||||||||
| Net proceeds from public offering | ||||||||||||
| Proceeds from new debt, net of costs | ||||||||||||
| Repayment of notes payable, inclusive of costs | ( | ) | ( | ) | ||||||||
| Payment of payroll taxes upon vesting of PSUs, RSUs and exercise of stock options | ( | ) | ( | ) | ( | ) | ||||||
| Proceeds from exercise of stock options | ||||||||||||
| Payment of debt issuance costs | ( | ) | ||||||||||
| Proceeds from public offering of common stock, net of offering costs | ||||||||||||
| NET CASH (USED IN) PROVIDED BY FINANCING ACTIVITIES | ( | ) | ||||||||||
| NET CHANGE IN CASH AND CASH EQUIVALENTS | ( | ) | ( | ) | ||||||||
| CASH AND CASH EQUIVALENTS, beginning of period | ||||||||||||
| CASH AND CASH EQUIVALENTS, end of period | $ | $ | $ | |||||||||
| SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | ||||||||||||
| Cash paid for income taxes | $ | $ | $ | |||||||||
| Cash paid for interest | $ | $ | $ | |||||||||
| SUPPLEMENTAL DISCLOSURES OF NON-CASH INVESTING AND FINANCING ACTIVITIES: | ||||||||||||
| Accrual of milestone payment | $ | $ | $ | |||||||||
| Reclassification of deferred financing costs | $ | $ | $ | |||||||||
| Accrual of exit fee related to Oaktree Loan | $ | $ | $ | |||||||||
| Purchase of property, plant and equipment included in accounts payable and accrued expenses | $ | $ | $ | |||||||||
| Change in right-of-use assets for operating lease obligations assumptions | $ | $ | ( | ) | $ | |||||||
| Right-of-use assets obtained in exchange for new operating lease obligations | $ | $ | $ | |||||||||
| Reclass of deferred commitment fees from prepaid expenses into debt issuance costs | $ | $ | $ | |||||||||
| Insurance premium financed | $ | $ | $ | |||||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-10 |
HARROW, INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. ORGANIZATION
Harrow, Inc. (together with its consolidated subsidiaries, unless the context indicates or otherwise requires, the “Company” or “Harrow”) is a leading eyecare pharmaceutical company engaged in the discovery, development, and commercialization of innovative ophthalmic pharmaceutical products for the U.S. market. Harrow helps U.S. eyecare professionals preserve the gift of sight by making its comprehensive portfolio of prescription and non-prescription pharmaceutical products accessible and affordable to millions of Americans each year. The Company owns commercial rights to one of the largest portfolios of branded ophthalmic pharmaceutical products in the U.S. all of which are marketed under its Harrow name. The Company also owns and operates ImprimisRx, one of the nation’s leading ophthalmology-focused pharmaceutical-compounding businesses.
Effective September 29, 2023, the Company changed its corporate name from Harrow Health, Inc. to Harrow, Inc. pursuant to a Certificate of Amendment to the Company’s Amended and Restated Certificate of Incorporation filed with the Secretary of State of the State of Delaware.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
Harrow has prepared the accompanying consolidated financial statements in accordance with accounting principles generally accepted in the U.S. (“GAAP”). The accompanying consolidated financial statements include the accounts of the Company and its wholly owned and majority-owned subsidiaries.
Harrow
consolidates entities in which it has a controlling financial interest. The Company assesses control under the variable interest entity
(“VIE”) model to determine whether the Company is the primary beneficiary of that entity. The Company consolidates
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and judgments that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and reported amounts of revenues and expenses during the reporting periods. Significant estimates made by management are, among others, allowance for credit losses, variable consideration determined based on accruals for chargebacks, administrative fees and rebates, government rebates, returns and other allowances, renewal periods and discount rates for leases, realizability of inventories, recoverability of investments, realizability of deferred tax assets, recoverability of long-lived assets and goodwill, valuations and purchase price allocations related to business combinations and asset acquisitions, fair value of loans payable, and valuation of stock-based transactions with employees and non-employees. Actual results could differ from those estimates.
Risks and Uncertainties
The Company is subject to certain regulatory standards, approvals, guidelines and inspections which could impact the Company’s ability to make, dispense, and sell certain products or subject the Company to enforcement actions. The Company’s 503B facility was inspected in 2024. The Company has been in discussions with the U.S. Food and Drug Administration (“FDA”) regarding the 2024 and other past FDA inspections of its 503B facility and has developed action plans to address observations made by FDA during these inspections and has communicated those plans to FDA. If the Company was required to cease compounding and selling certain products as a result of regulatory guidelines or inspections, it could have a material impact on the Company’s financial condition, liquidity and results of operations.
| F-11 |
Credit Losses
The Company estimates and records a provision for its expected credit losses related to its financial instruments, including its trade receivables. Management considers historical collection rates, the current financial status of the Company’s customers, macroeconomic factors, and other industry-specific factors when evaluating current expected credit losses. Forward-looking information is also considered in the evaluation of current expected credit losses. However, because of the short time to the expected receipt of accounts receivable, management believes that the carrying value, net of expected losses, approximates fair value and therefore, relies more on historical and current analysis of such financial instruments, including its trade receivables.
To determine the provision for credit losses for accounts receivable, the Company has disaggregated its accounts receivable by class of customer at the business component level, as management determined that risk profile of the Company’s customers is consistent based on the type and industry in which they operate, mainly in the pharmaceuticals industry. Each business component is analyzed for estimated credit losses individually. In doing so, the Company establishes a historical loss matrix, based on the previous collections of accounts receivable by the age of such receivables, and evaluates the current and forecasted financial position of its customers, as available. Further, the Company considers macroeconomic factors and the status of the pharmaceuticals industry to estimate if there are current expected credit losses within its trade receivables based on the trends of the Company’s expectation of the future status of such economic and industry-specific factors. Also, specific allowance amounts are established based on review of outstanding invoices to record the appropriate provision for customers that have a higher probability of default.
Accounts
receivable at December 31, 2025 and 2024 are net of allowances for credit losses of $
| Balance at January 1, 2023 | $ | |||
| Change in expected credit losses | ||||
| Write-offs, net of recoveries | ( | ) | ||
| Balance at December 31, 2023 | ||||
| Change in expected credit losses | ||||
| Write-offs, net of recoveries | ( | ) | ||
| Balance at December 31, 2024 | ||||
| Change in expected credit losses | ||||
| Write-offs, net of recoveries | ( | ) | ||
| Balance at December 31, 2025 | $ |
Business Combinations and Asset Acquisitions
The Company evaluates acquisitions of assets and other similar transactions to assess whether the transaction should be accounted for as a business combination or asset acquisition by first applying a screen to determine if substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or group of similar identifiable assets. If the screen is met, the transaction is accounted for as an asset acquisition. If the screen is not met, further determination is required as to whether the Company has acquired inputs, process, and output, which would meet the requirements of a business. If determined to be a business combination, the Company accounts for the transaction under the acquisition method of accounting as indicated in the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification (“ASC”).
ASC 805, Business Combinations, requires the acquiring entity in a business combination to recognize the fair value of all assets acquired, liabilities assumed, and any non-controlling interest in the acquiree and establishes the acquisition date as the fair value measurement point. Accordingly, the Company recognizes assets acquired and liabilities assumed in business combinations, including any contingent assets and liabilities, and any non-controlling interest in the acquiree based on the fair value estimates as of the date of acquisition. The Company recognizes and measures goodwill as of the acquisition date, as the excess of the fair value of the consideration paid over the fair value of the identified net assets acquired.
| F-12 |
The consideration for the Company’s business acquisitions may include future payments that are contingent upon the occurrence of a particular event or events. The obligations for such contingent consideration payments are recorded at fair value on the acquisition date. The contingent consideration obligations are then evaluated each reporting period. Changes in the fair value of contingent consideration, other than changes due to payments, would be recognized as a gain or loss and recorded in the consolidated statement of operations.
If determined to be an asset acquisition, the Company accounts for the transaction under ASC 805-50, Business Combinations – Related Issues, which requires the acquiring entity in an asset acquisition to recognize assets acquired and liabilities assumed based on the cost to the acquiring entity or a relative fair value basis, which includes transaction costs in addition to consideration given. No gain or loss is recognized as of the date of acquisition unless the fair value of non-cash assets given as consideration differs from the assets’ carrying amounts on the acquiring entity’s financial statements. Consideration transferred that is non-cash will be measured based on either the cost (which shall be measured based on the fair value of the consideration given) or the fair value of the assets acquired, and liabilities assumed, whichever is more clearly evident and more reliably measurable. The obligation for contingent consideration payments is recorded when the contingency is resolved and is probable and reasonably estimable. Contingent consideration recognized is included in the initial cost of the assets acquired and any subsequent changes in the recorded amount of contingent consideration are recognized as an adjustment to the cost basis of the acquired assets and allocated to the acquired assets based on the relative fair value at the date of acquisition. Goodwill is not recognized in an asset acquisition and any excess consideration transferred over the fair value of the net assets acquired is allocated to the identifiable assets based on relative fair values.
Noncontrolling Interests
The
Company recognizes any noncontrolling interest as a separate line item in equity in the consolidated financial statements. A noncontrolling
interest represents the portion of equity ownership in a less-than-wholly-owned subsidiary not attributable to the Company.
The Company provides in the consolidated statements of stockholders’ equity a reconciliation at the beginning and the end of the period of the carrying amount of total equity, equity attributable to the parent, and equity attributable to the noncontrolling interests that separately discloses:
| 1. | net income or loss; | |
| 2. | transactions with owners acting in their capacity as owners, showing separately contributions from and distributions to owners; and | |
| 3. | each component of other income or loss. |
The noncontrolling interests in the consolidated balance sheets as of December 31, 2025 and 2024, relate to consolidated subsidiaries for which the Company does not own 100% of the equity interests, and that no longer have active operations, assets and related financial activity.
Revenue Recognition and Deferred Revenue
The Company recognizes revenue at the time of transfer of promised goods or services to customers in an amount that reflects the consideration to which the Company expects to be entitled in exchange for those goods or services (see Note 3).
| F-13 |
Cost of Sales
Cost of sales includes direct and indirect costs to manufacture formulations and other products sold, including APIs, personnel costs, packaging, storage, royalties, shipping and handling costs, depreciation and amortization of certain intangible assets and the write-off of obsolete inventory.
Research and Development
Research and development (“R&D”) expenses consist of expenses incurred in performing research and development activities, including salaries and benefits, other overhead expenses, and costs related to clinical trials, contract services and outsourced contracts. The Company expenses all costs related to R&D as they are incurred.
Upfront and milestone payments related to the acquisition and licensing of technology for drug and product candidates that are not yet approved by the FDA are considered acquisition of in-process R&D and expensed as R&D in the period in which the expense occurs.
Advertising Expense
Advertising
costs are expensed as incurred and are included in selling, general and administrative expense in the consolidated statements of operations.
Advertising expense was $
Debt Issuance Costs and Debt Discount
Debt issuance costs and the debt discount are recorded net of notes payable in the consolidated balance sheets. Amortization of debt issuance costs and the debt discount is calculated using the effective interest method over the term of the related debt and is recorded in interest expense in the accompanying consolidated statements of operations.
Intellectual Property
The costs of acquiring intellectual property rights to be used in the research and development process, including licensing fees and milestone payments, are charged to research and development expense as incurred in situations where the Company has not identified an alternative future use for the acquired rights, and are capitalized in situations where the Company has identified an alternative future use for the acquired rights. Patents and trademarks are recorded at cost and capitalized at a time when the future economic benefits of such patents and trademarks become more certain (see “Intangible Assets, including Goodwill” below). If not capitalized, the costs are expensed as incurred.
Income Taxes
As part of the process of preparing the Company’s consolidated financial statements, the Company must estimate the actual tax assets and liabilities and assess permanent and temporary differences that result from differing treatment of items for tax and accounting purposes. The temporary differences result in deferred tax assets and liabilities, which are included within the consolidated balance sheets. The Company must assess the likelihood that the deferred tax assets will be recovered from future taxable income and, to the extent the Company believes that recovery is not more likely than not, a valuation allowance must be established which reduces the amount of deferred tax assets recorded on the consolidated balance sheets. To the extent the Company establishes a valuation allowance or increase or decrease this allowance in a period, the impact will be included in income tax expense in the consolidated statements of operations.
The
Company accounts for income taxes under the provisions of ASC 740, Income Taxes. As of December 31, 2025 and 2024, there was $
| F-14 |
Cash and Cash Equivalents
Cash equivalents include short-term, highly liquid investments with maturities of three months or less at the time of acquisition.
Concentrations of Credit Risk
The
Company places its cash with financial institutions deemed by management to be of high credit quality. The Federal Deposit Insurance
Corporation (“FDIC”) provides basic deposit coverage with limits up to $
Accounts Receivable
Accounts
receivable is stated net of allowances for credit losses and contractual adjustments. The accounts receivable balance primarily includes
amounts due from customers the Company has invoiced or from third-party providers (e.g., insurance companies and governmental agencies),
but for which payment has not been received. The Company’s gross product revenues are subject to a variety of contractual deductions,
which generally are estimated and recorded in the same period that the revenues are recognized. These deductions represent estimates
of the related obligations and, as such, knowledge and judgment are required when estimating the impact of these revenue deductions on
gross sales for a reporting period. Accounts receivable at December 31, 2025 are presented net of allowances for credit losses of $
Inventories
Inventories are stated at the lower of cost or net realizable value. Cost is determined on a first-in, first-out basis. The Company evaluates the carrying value of inventories on a regular basis, based on the price expected to be obtained for products in their respective markets compared with historical cost. Write-downs of inventories are considered to be permanent reductions in the cost basis of inventories.
The Company also regularly evaluates its inventories for excess quantities and obsolescence (expiration), taking into account such factors as historical and anticipated future sales or use in production compared to quantities on hand and the remaining shelf life of products and APIs on hand. The Company establishes reserves for excess and obsolete inventories as required based on its analyses.
Investment in Surface Ophthalmics, Inc. – Related Party
The
Company owns common shares of Surface Ophthalmics, Inc. (“Surface”), representing approximately
Under the equity method, the Company recognizes its proportionate share of Surface’s net earnings or losses in its consolidated statements of operations and adjusts the carrying amount of its investment accordingly. The Company’s share of earnings or losses is based on its ownership interest in Surface, and intra-entity profits and losses are eliminated.
During
the year ended December 31, 2021, the Company reduced the carrying value of its investment in Surface to zero after recognizing cumulative
equity method losses equal to its investment balance of $
| F-15 |
The following table summarizes the Company’s investment in Surface as of December 31, 2025 and 2024:
| Cost Basis | Share of Equity Method Losses | Net Carrying value | ||||||||||
| Common stock | $ | $ | ( | ) | $ | |||||||
See Note 6 for more information and related party disclosure regarding Surface.
Property, Plant and Equipment
Property,
plant and equipment is stated at cost less accumulated depreciation and amortization. Depreciation and amortization is calculated using
the straight-line method over the estimated useful life of the asset. Leasehold improvements and finance lease equipment are amortized
over the estimated useful life or remaining lease term, whichever is shorter. Computer hardware and furniture and equipment are depreciated
over three to
Capitalized Software Costs
The
Company capitalizes certain costs related to the development of internal-use software. Costs incurred during the application development
phase are capitalized only when the Company believes it is probable the development will result in new or additional functionality. The
types of costs capitalized during the application development phase include consulting fees for third-party developers working on these
projects. Costs related to the preliminary project stage and post-implementation activities are expensed as incurred. Internal-use software
is amortized on a straight-line basis over the estimated useful life of the asset, which ranges from two to
Intangible Assets, including Goodwill
Patents and trademarks are recorded at cost and capitalized at a time when the future economic benefits of such patents and trademarks become more certain. At that time, the Company capitalizes third-party legal costs and filing fees associated with obtaining and successfully prosecuting claims related to its patents and trademarks. Once the patents have been issued, the Company amortizes these costs over the shorter of the legal life of the patent or its estimated economic life using the straight-line method. Acquired product rights, including new drug applications (“NDAs”), are amortized over their estimated useful lives based on a straight-line method. Trademarks are an indefinite-lived intangible asset and are assessed for impairment based on future projected cash flows as further described below.
We review our goodwill and indefinite-lived intangible assets for impairment annually as of January 1 of each year and when an event or a change in circumstances indicates the fair value of a reporting unit for goodwill or individual asset for indefinite-lived intangible assets may be below its carrying amount. We first perform a qualitative assessment to determine whether it is more likely than not that the fair value of a reporting unit or individual asset is less than its carrying amount. In performing this assessment, we consider factors such as macroeconomic conditions, industry and market trends, cost factors, overall financial performance, changes in management or strategy, and other relevant events or circumstances. If, after evaluating these factors, we conclude that it is more likely than not that the fair value exceeds its carrying amount, no further analysis is required.
If the qualitative assessment indicates that a quantitative impairment test is necessary, we estimate the fair value of the reporting unit or individual asset and compare it to its carrying amount. Fair value is determined using a discounted cash flow analysis or other appropriate valuation techniques.
| F-16 |
If the fair value of the reporting unit or individual asset exceeds its carrying amount, the intangible asset is not considered impaired. If the carrying amount of the reporting unit exceeds its fair value, we recognize a goodwill impairment loss for the amount of the excess, limited to the total amount of goodwill allocated to that reporting unit. Indefinite-lived intangible asset impairment is measured by comparing the individual asset’s estimated fair value with its carrying amount, with any excess of carrying amount over fair value recognized as an impairment loss.
As
a result of our assessments in 2025 and 2024, we concluded that goodwill and indefinite-lived intangible assets are
Impairment of Other Long-Lived Assets
Other long-lived assets, such as property, plant and equipment, purchased intangibles subject to amortization and patents and trademarks, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Such circumstances could include, but are not limited to (1) a significant decrease in the market value of an asset, (2) a significant adverse change in the extent or manner in which an asset is used, or (3) an accumulation of costs significantly in excess of the amount originally expected for the acquisition of an asset. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated undiscounted future cash flows, an impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. The fair value of the asset is based on the discounted value of its estimated future cash flows. Assets to be disposed of would be separately presented in the consolidated balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated. The assets and liabilities of a disposal group classified as held-for-sale would be presented separately in the appropriate asset and liability sections of the consolidated balance sheet, if material.
Leases
At the inception of a contract the Company determines if the arrangement is, or contains, a lease. Operating lease right-of-use (“ROU”) assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent its obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term, discounted using the Company’s incremental borrowing rate of the debt outstanding. Lease expense is recognized on a straight-line basis over the lease term.
The Company has made certain accounting policy elections whereby it (i) does not recognize ROU assets or lease liabilities for short-term leases (those with original terms of 12-months of less) and (ii) combines lease and non-lease elements of its operating leases as a single lease component. As of December 31, 2025 and 2024, the Company did not have any finance leases.
Fair Value Measurements
Fair value measurements are determined based on the assumptions that market participants would use in pricing an asset or liability. GAAP establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. The established fair value hierarchy prioritizes the use of inputs used in valuation methodologies into the following three levels:
| ● | Level 1: Applies to assets or liabilities for which there are quoted prices (unadjusted) for identical assets or liabilities in active markets. A quoted price in an active market provides the most reliable evidence of fair value and must be used to measure fair value whenever available. |
| ● | Level 2: Applies to assets or liabilities for which there are significant other observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| ● | Level 3: Applies to assets or liabilities for which there are significant unobservable inputs that reflect a reporting entity’s own assumptions about the assumptions that market participants would use in pricing an asset or liability. For example, Level 3 inputs would relate to forecasts of future earnings and cash flows used in a discounted future cash flows method. |
| F-17 |
The 2030 Notes (as defined in Note 13) are carried at face value less unamortized debt issuance costs. The Company fully repaid principal balances under the Oaktree Loan (as defined in Note 13) and the 2026 and 2027 Notes (each as defined in Note 13) in September 2025. The Company’s 2026 Notes were carried at face value, including the unamortized premium, less unamortized debt issuance costs, the 2027 Notes were carried at face value less unamortized debt issuance costs, and the Oaktree Loan was carried at face value less the original issue discount and unamortized debt issuance costs on the consolidated balance sheets and the Company presents fair value for disclosure purposes only. The 2026 Notes and the 2027 Notes were classified as Level 1 instruments as the fair value was determined using quoted market prices in active markets for the same securities. The 2030 Notes are classified as Level 1 instruments. The Oaktree Loan was classified as a Level 2 instrument as the fair value is determined through an income approach that considers collateral coverage, yield calibration, yield analysis and any adjustments to implied yield associated with the Company’s fundamental measures.
The following table presents the estimated fair values and the carrying values:
| December 31, | ||||||||||||||||
| 2025 | 2024 | |||||||||||||||
| Carrying Value | Fair Value | Carrying Value | Fair Value | |||||||||||||
| 2030 Notes | $ | $ | ||||||||||||||
| 2026 Notes | $ | $ | $ | $ | ||||||||||||
| 2027 Notes | $ | $ | $ | $ | ||||||||||||
| Oaktree Loan | $ | $ | $ | $ | ||||||||||||
The Company’s other financial instruments include cash and cash equivalents, accounts receivable, accounts payable and accrued expenses, accrued payroll and related liabilities, deferred revenue and customer deposits and operating lease liabilities. The carrying amount of these financial instruments, except for operating lease liabilities, approximates fair value due to the short-term maturities of these instruments. Based on borrowing rates currently available to the Company, the carrying values of the operating lease liabilities approximate their respective fair values.
Stock-Based Compensation
All stock-based payments to employees, directors and consultants, including grants of stock options, warrants, restricted stock units (“RSUs”), performance stock units (“PSUs) and restricted stock, are recognized in the consolidated financial statements based upon their estimated fair values. The Company uses the Black-Scholes-Merton option pricing model and Monte Carlo simulation model to estimate the fair value of stock-based awards. The estimated fair value is determined at the date of grant. The financial statement effect of forfeitures is estimated at the time of grant and revised, if necessary, if the actual effect differs from those estimates. The Company provides newly issued shares of common stock to satisfy the exercise and vesting for stock-based compensation awards.
Basic net loss per common share is computed by dividing net loss attributable to Harrow, Inc. for the year by the weighted average number of common shares outstanding during the year. Diluted net loss per share is computed by dividing the net loss attributable to Harrow, Inc. for the year by the weighted average number of common and common equivalent shares, such as stock options, RSUs, PSUs, and warrants, outstanding during the year.
Common stock equivalents (using the treasury stock or “if converted” method) from stock options, unvested RSUs, and unvested PSUs were , , and at December 31, 2025, 2024 and 2023, respectively, and are excluded in the calculation of diluted net loss per share for the periods presented, because the effect is anti-dilutive for that time period. Included in the basic and diluted net loss per share calculation were RSUs awarded to directors that had vested, but the issuance and delivery of the shares are deferred until the director resigns. The number of shares underlying vested but unissued RSUs at December 31, 2025, 2024 and 2023 was , and , respectively.
| F-18 |
Reclassifications
The December 31, 2024 balances in the schedule of deferred tax assets and liabilities in Note 16 - Income Taxes have been reclassified to conform to current year presentation.
Recently Adopted Accounting Pronouncements
In December 2023, the FASB issued Accounting Standards Update (“ASU”) 2023-09, Income Taxes (Topic 740) - Improvements to Income Tax Disclosures, which enhances the disclosures required for income taxes in the Company’s annual consolidated financial statements. Notably, this ASU requires entities to disclose specific categories in the effective tax rate reconciliation and provide additional information for reconciling items that meet a quantitative threshold. The Company adopted ASU 2023-09 as of December 31, 2025. The adoption did not impact the Company’s consolidated financial results of operations, financial position or cash flows.
Accounting Guidance Issued but Not Adopted at December 31, 2025
In October 2023, the FASB issued ASU 2023-06, Disclosure Improvements—Codification Amendments in Response to the SEC’s Disclosure Update and Simplification Initiative. This ASU modifies the disclosure or presentation requirements of a variety of topics in the codification by aligning them with the SEC’s regulations. The amendments to the various topics should be applied prospectively, and the effective date for the Company for each amendment will be determined based on the effective date of the SEC’s removal of the related disclosure from Regulation S-X or Regulation S-K. If the SEC has not removed the applicable requirement by June 30, 2027, then the related amendment in ASU 2023-06 will be removed from the codification and will not become effective. Early adoption of this ASU is prohibited. The Company does not expect the amendments in this ASU to have a material impact on the disclosures or presentation in its consolidated financial statements.
In November 2024, the FASB issued ASU 2024-03, Income Statement – Reporting Comprehensive Income – Expense Disaggregation Disclosures, to improve the disclosures by a public business entity about the types of expenses in commonly presented expense captions. This ASU is effective for annual reporting periods beginning after December 15, 2026, and interim reporting periods beginning after December 15, 2027, with early adoption permitted. The Company is currently evaluating the impact of ASU 2024-03 on its consolidated financial statements.
NOTE 3. REVENUES
The Company accounts for contracts with customers in accordance with ASC 606, Revenues from Contracts with Customers. The Company has two primary streams of revenue: (1) product revenues, including revenue recognized from sales of products through its pharmacy and outsourcing facility and sales of branded products to wholesalers through a third-party logistics (“3PL”) partner, and (2) revenue recognized from intellectual property licenses.
Product Revenues
The Company sells prescription medications directly through its pharmacy, outsourcing facility and 3PL partner. Revenue from the Company’s pharmacy services includes: (i) the portion of the price the client pays directly to the Company, net of any volume-related or other discounts paid back to the client, (ii) the price paid to the Company by individuals, and (iii) customer copayments made directly to the pharmacy network. Sales taxes are not included in revenue. Following the core principles of ASC 606, the Company has identified the following:
| 1. | Identify the contract(s) with a customer: A contract is deemed to exist when the customer places an order through receipt of a prescription, via an online order or via receipt of a purchase order from a customer. For branded products, orders are received through the Company’s 3PL partner, and the customer takes title of the products via formal purchase orders placed and fulfilled. |
| 2. | Identify the performance obligations in the contract: Obligations for fulfillment of the Company’s contracts consist of delivering the product to customers at their specified destination. For shipping and handling activities under ASC 606, if the customer takes control of the goods after shipment, shipping and handling activities would always be considered a fulfillment activity and not treated as a separate performance obligation. If the customer takes control of the goods before shipment, entities must make an accounting policy election to treat shipping and handling activities as either a fulfillment cost or as a separate performance obligation. The Company has elected to treat its shipping and handling activities as a fulfillment cost. |
| F-19 |
| 3. | Determine the transaction price: The transaction price is based on an amount that reflects the consideration to which the Company expects to be entitled, net of accruals for estimated rebates, wholesaler chargebacks, discounts, copay assistance and other deductions (collectively, sales deductions) and an estimate for returns and replacements established at the time of sale. The Company utilizes the services of a third-party professional services firm to estimate rebates and chargebacks associated with sales of its branded products. The transfer of promised goods is satisfied within a year, and therefore there are no significant financing components. There is no non-cash consideration related to product sales. |
| 4. | Allocate the transaction price to the performance obligations in the contract: Because there is only one performance obligation for product sales, no allocation is necessary. |
| 5. | Recognize revenue when (or as) the entity satisfies a performance obligation: Revenue from products is recognized upon transfer of control of a product to a customer. This generally occurs upon shipment unless contractual terms with a customer state that transfer of control occurs at delivery. |
Variable Consideration
Sales of branded pharmaceutical products are subject to variable consideration due to chargebacks, government rebates, returns, administrative fees, co-pay assistance and other rebates, and prompt pay discounts. Estimates for these elements of variable consideration require significant judgment.
Chargebacks
Chargebacks, primarily from distributors and wholesalers, result from arrangements with indirect customers establishing prices for products which the indirect customer purchases through a wholesaler. Alternatively, the Company may pre-authorize wholesalers to offer specified contract pricing to other indirect customers. Under either arrangement, the Company provides a chargeback credit to the wholesaler for any difference between the contracted price with the indirect customer and the wholesaler’s invoice price, typically Wholesale Acquisition Cost (“WAC”).
Prior period chargebacks claimed by wholesalers are analyzed to determine the actual net price per package (“NPP”) for each product. This calculation is performed by product, by wholesaler. NPPs can be affected by several factors such as:
| ● | Changes in customer mix |
| ● | Changes in negotiated terms with customers |
| ● | Changes in the volume of off-contract purchases |
| ● | Changes in WAC |
As necessary, NPPs are adjusted based on anticipated changes in the factors above.
The difference between NPP and WAC is recorded as a reduction in both gross revenues in the consolidated statements of operations and accounts receivable in the consolidated balance sheets, at the time revenue is recognized from the product sale. The Company continually monitors chargeback activity and adjusts NPPs when the Company believes that actual selling prices will differ from current NPPs.
Government Rebates
Government rebates reserve consists of estimated payments due to governmental agencies for utilization of the Company’s products by beneficiaries under such governmental programs. The two largest government programs are Medicaid and Medicare.
| F-20 |
The Company participates in the Medicaid Drug Rebate Program and pays rebates to the states related to Medicaid beneficiary utilization of the Company’s products. Medicaid rebates are billed within 60-90 days of the end of the quarter in which the product was dispensed to a Medicaid beneficiary. Medicaid rebate amounts per product unit are established by law, based on the Average Manufacturer Price (“AMP”), which is reported on a monthly and quarterly basis, and, in the case of branded products, best price, which is reported on a quarterly basis. Medicaid reserves are based on expected claims from state Medicaid programs. Estimates for expected claims are driven by patient usage, sales mix, calculated AMP or best price, as well as inventory in the distribution channel that will be subject to a Medicaid rebate. As a result of the delay between selling the products, dispensing the products and rebate billing, the Medicaid rebate reserve includes both an estimate of outstanding claims for end-customer sales that have occurred but for which the related claim has not been billed, as well as an estimate for future claims that will be made when inventory in the distribution channel is sold through to plan participants. Many of the Company’s branded products are also covered under Medicare. The Company participates in the Coverage Gap Discount Program in order for its branded products to be covered by Medicare Part D and must provide a rebate for any products sold under NDAs dispensed to Medicare Part D beneficiaries while the beneficiaries are in the Coverage Gap phase of the benefit. This applies to all products sold under NDAs. Estimates for these discounts are based on historical experience with Medicare rebates for products. Medicare rebates are billed quarterly for drugs dispensed to Medicare beneficiaries in the prior quarter, which is typically 120 days after the product is shipped. As a result of the delay between selling the products, dispensing the products and rebate billing, Medicare rebate reserve includes both an estimate of outstanding claims for end-customer sales that have occurred but for which the related claim has not been billed, as well as an estimate for future claims that will be made when inventory in the distribution channel is sold through to Medicare Part D participants.
To evaluate the adequacy of the government rebate reserves, reserves are reviewed on a quarterly basis against actual claims data to ensure the liability is fairly stated. The Company continually monitors the government rebate reserve and adjusts estimates if it is expected that actual government rebates may differ from established accruals. Accruals for government rebates are recorded as a reduction to gross revenues in the consolidated statements of operations and as an increase to accrued rebates in the consolidated balance sheets.
Returns
A returns policy is in place that allows customers to return product within a specified period prior to and subsequent to the expiration date. Generally, product may be returned for a period beginning six months prior to its expiration date to up to one year after its expiration date. Product returns are settled through the issuance of a credit to the customer. The estimate for returns is based upon historical experience with actual returns. While such experience has allowed for reasonable estimation in the past, history may not always be an accurate indicator of future returns. The Company continually monitors estimates for returns and adjusts when it is expected that actual product returns may differ from the established accruals. Accruals for returns are recorded as a reduction to gross revenues in the consolidated statements of operations and as an increase to accrued expenses in the consolidated balance sheets.
Administrative Fees and Other Rebates
Administrative fees or rebates are offered to wholesalers, group purchasing organizations, and indirect customers. Fees and rebates are accrued, by product by wholesaler, at the time of sale based on contracted rates and NPP. To evaluate the adequacy of the administrative fee accruals, on-hand inventory counts are obtained from the wholesalers. The Company continually monitors administrative fee activity and adjusts accruals when it is expected that actual administrative fees may differ from the accruals. Accruals for administrative fees and other rebates are recorded as a reduction in both gross revenues in the consolidated statements of operations and accounts receivable or accrued expenses in the consolidated balance sheets.
Co-payment Assistance
Patients who meet certain eligibility requirements may receive co-payment assistance funded by the Company. The Company records contra-revenue for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators. An accrued liability is recorded and revenue is reduced on unredeemed co-payment assistance related to products for which control has been transferred to the customer.
| F-21 |
Prompt Payment Discounts
Sales discounts may be granted to customers for prompt payment. The reserve for prompt payment discounts is based on invoices outstanding. Based on past experience, it is assumed that all available discounts will be taken. Accruals for prompt payment discounts are recorded as a reduction in both gross revenues in the consolidated statements of operations and accounts receivable in the consolidated balance sheets.
The following table summarizes activity and ending balances of the Company’s variable consideration provisions in the consolidated financial statements for the years ended December 31, 2025, 2024 and 2023:
| Accruals for Chargebacks, Returns, and Other Allowances | ||||||||||||||||||||||||||||
| Chargebacks | Government Rebates | Returns | Administrative Fees and Other Rebates | Co-Pay Assistance | Prompt Pay Discounts | Total | ||||||||||||||||||||||
| Balance at December 31, 2022 (1) | $ | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||
| Accruals/Adjustments | ||||||||||||||||||||||||||||
| Credits Taken Against Reserve | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| Balance at December 31, 2023 (1) | ||||||||||||||||||||||||||||
| Accruals/Adjustments | ||||||||||||||||||||||||||||
| Credits Taken Against Reserve | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| Balance at December 31, 2024 (1) | ||||||||||||||||||||||||||||
| Accruals/Adjustments | ||||||||||||||||||||||||||||
| Credits Taken Against Reserve | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| Balance at December 31, 2025 (1) | $ | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||
| (1) |
Revenues From Transfer of Acquired Product Sales and Profits
During 2024 and 2023, the Company was a party to agreements whereby it purchased the exclusive commercial rights to assets associated with certain ophthalmic products from other pharmaceutical companies (the “Sellers”). During a temporary, transition period, the Sellers continue to manufacture and market these products and transfer the net profit from the sale of the products to the Company. The revenue recognized by the Company from the transfer of net profit was recognized at the time profit from the product sales were calculated by the Sellers and confirmed by the Company, typically on a monthly basis, at which point there is no future performance obligation required by the Company and no consequential continuing involvement on the Company’s part to recognize the associated revenue. On a quarterly basis, the Sellers invoice the Company for all credits and reimbursements (“Chargebacks”) made to customers related to the products. The Company uses historical actual experience to estimate Chargebacks associated with the net sales and profit transferred. The estimated Chargebacks are recorded as a reduction in revenues from transfer of acquired product sales and profits in the Company’s consolidated statements of operations, and recorded as a reduction to accounts receivable in the consolidated balance sheets, at the time the revenue is recognized.
| F-22 |
Other Revenue: Intellectual Property License and Related Arrangements
The Company currently holds five intellectual property licenses and related agreements pursuant to which the Company has agreed to license or sell to a customer with the right to access the Company’s intellectual property. License arrangements may consist of non-refundable upfront license fees, data transfer fees, research reimbursement payments, exclusive license rights to patented or patent pending compounds, technology access fees, and various performance or sales milestones. These arrangements can be multiple-element arrangements, the revenue of which is recognized at the point in time that the performance obligation is met.
Non-refundable fees that are not contingent on any future performance by the Company and require no consequential continuing involvement on the part of the Company are recognized as revenue when the license term commences and the licensed data, technology, compounded drug preparation and/or other deliverables are delivered. Such deliverables may include physical quantities of compounded drug preparations, design of the compounded drug preparations and structure-activity relationships, the conceptual framework and mechanism of action, and rights to the patents or patent applications for such compounded drug preparations. The Company defers recognition of non-refundable fees if it has continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee and that are separate and independent of the Company’s performance under the other elements of the arrangement. In addition, if the Company’s continued involvement is required, through research and development services that are related to its proprietary know-how and expertise of the delivered technology or can only be performed by the Company, then such non-refundable fees are deferred and recognized over the period of continuing involvement. Guaranteed minimum annual royalties are recognized on a straight-line basis over the applicable term.
Deferred
revenue and customer deposits at December 31, 2025 and 2024, were $
NOTE 4. RECENT PRODUCT ACQUISITIONS AND LICENSES
FDA Approved Product Acquisitions
In recent years, the Company has acquired commercial and product rights to various FDA approved ophthalmic medications and products through asset purchase, licenses, supply and/or other related agreements. In general, in exchange for product and commercial rights these agreements provide the counterparties with certain upfront and contingent milestone payments typically related to certain annual sales amounts and manufacturing events, and in certain cases, per unit transfer prices and royalties on sales of some of the products.
During
the years ended December 31, 2025, 2024 and 2023, $
BYOOVIZ® and OPUVIZTM – Commercialization Agreement
In
July 2025, the Company entered into a development and commercialization agreement (the “Samsung Agreement”) with Samsung
Bioepis Co., Ltd. (“Samsung”). Under the terms of the Samsung Agreement, following completion of the transition of commercial
rights from Biogen, Inc. back to Samsung, Samsung will develop, manufacture, and supply BYOOVIZ (ranibizumab-nuna) and OPUVIZ (aflibercept-yszy)
(individually, a “Product” and together, the “Products”) for Harrow to commercialize in the U.S. market (the
“Rights”). The Company accounted for the transaction as an acquisition of assets and capitalized the one-time upfront payment
of $
| F-23 |
Contingent consideration related to the transaction will be recognized only when the contingency is resolved and the consideration is considered payable.
Acquisition of Commercial Rights to BYQLOVITM
In
June 2025, the Company announced that it had entered into a license and supply agreement (the “Formosa Agreement”) with Formosa
Pharmaceuticals, Inc. (“Formosa”). Under the terms of the Formosa Agreement,
Acquisition of VEVYE® U.S. and Canadian Commercial Rights
In
July 2023, the Company acquired commercial rights of VEVYE (cyclosporine ophthalmic solution)
The
Company accounted for the VEVYE Acquisition as an acquisition of assets and capitalized the initial payments of $
During
the year ended December 31, 2025, the Company capitalized a commercial milestone of $
Acquisition of Certain U.S. and Canadian Commercial Rights to Santen and Eyevance Products
In July 2023, the Company entered into an Asset Purchase Agreement with Eyevance Pharmaceuticals, LLC and a License Agreement with Santen S.A.S. (collectively, the “Santen Agreements”), each a subsidiary of Santen Pharmaceuticals Co., Ltd. (collectively, “Santen”). Pursuant to the Santen Agreements, the Company acquired the exclusive commercial rights to assets associated with the following ophthalmic products (collectively, the “Santen Products”): FLAREX, NATACYN, ZERVIATE, VERKAZIA and FRESHKOTE in the U.S., and VERKAZIA and CATIONORM PLUS in Canada.
The transactions pursuant to the Santen Agreements are referred to in these notes as the “Santen Products Acquisition.”
Under
the terms of the Santen Agreements, the Company made an initial one-time payment of $
The assets acquired in the Santen Products Acquisition are identifiable intangible asset groups in similar asset classes and all directly related to the product NDAs and marketing authorizations acquired. The developed technology is within one major intangible asset class. No workforce/employees were included in the Santen Products Acquisition and the Company is required to utilize its own business inputs/processes to transfer and commercialize the Santen Products.
| F-24 |
The
Company incurred $
Acquisition of ILEVRO, NEVANAC, VIGAMOX, MAXIDEX, and TRIESENCE
In December 2022, the Company entered into an Asset Purchase Agreement (the “NVS 5 APA”) with Novartis Technology, LLC and Novartis Innovative Therapies AG (together, “Novartis”), pursuant to which the Company agreed to purchase from Novartis the exclusive commercial rights to assets associated with the following ophthalmic products (collectively the “NVS 5 Products”) in the U.S. (the “NVS 5 Acquisition”): ILEVRO, NEVANAC, VIGAMOX, MAXIDEX, and TRIESENCE.
Under
the terms of the NVS 5 APA, the Company made a one-time payment of $
The assets acquired in the NVS 5 Acquisition are identifiable intangible asset groups in similar asset classes and all directly related to the five product NDAs acquired. The developed technology is within one major intangible asset class. No workforce/employees were included in the NVS 5 Acquisition, and the Company is required to utilize its own business inputs/processes to transfer and commercialize the NVS 5 Products and NDAs.
The
Company incurred $
NOTE 5. INVESTMENT IN MELT PHARMACEUTICALS, INC. AND AGREEMENTS – RELATED PARTY TRANSACTIONS
In December 2018, the Company entered into an asset purchase agreement (the “Melt APA”) with Melt Pharmaceuticals, Inc. (“Melt”). Pursuant to the terms of the Melt APA, Melt was assigned certain intellectual property and related rights from the Company to develop, formulate, make, sell, and sub-license certain Company conscious sedation and analgesia related formulations (collectively, the “Melt Products”). Under the terms of the Melt APA, Melt was required to make mid-single digit royalty payments to the Company on net sales of the Melt Products while any patent rights remain outstanding, as well as other conditions.
In
connection with the settlement of a Note receivable from Melt of $
| F-25 |
In
accordance with ASC 323, Investments – Equity Method and Joint Ventures, the carrying amount of the note receivable and
other investments in Melt have been reduced to
Acquisition of Remaining Interests in Melt Pharmaceuticals, Inc. - Related Party
In September 2025, the Company entered into an Agreement and Plan of Merger (the “Merger Agreement”) by and among the Company, Harrow Acquisition Sub, Inc., a wholly owned subsidiary of the Company, Melt Pharmaceuticals, Inc. (“Melt”), and D. Brad Osborne, as stockholder representative. The transaction closed on November 17, 2025.
Under
the terms of the Merger Agreement and related milestone payment agreement, the Company agreed to acquire the remaining equity interests
of Melt in exchange for an initial cash payment of approximately $
| ● | Upon FDA approval of MELT-300,
the Company shall pay an aggregate amount in cash of approximately $ | |
| ● | Upon receipt of pass-through status awarded and J-Code (or any other similar designation) issued by the Center for Medicare & Medicaid Services for MELT-300 the Company shall issue an aggregate of approximately shares of the Company’s common stock. | |
| ● |
The regulatory and commercial milestones must be achieved on or before December 31, 2035.
The
Company accounted for the Melt transaction as an asset acquisition and expensed the one-time upfront consideration of $
NOTE 6. INVESTMENT IN SURFACE OPHTHALMICS, INC. AND AGREEMENTS - RELATED PARTY TRANSACTIONS
The Company entered into an asset purchase and license agreement with Surface in 2017 and amended it in April 2018 (the “Surface License Agreements”). Pursuant to the terms of the Surface License Agreements, the Company assigned and licensed to Surface certain intellectual property and related rights associated with Surface’s drug candidates (collectively, the “Surface Products”). Surface is required to make mid-single digit royalty payments to the Company on net sales of the Surface Products while any patent rights remain outstanding The Company has not received any royalties related to the Surface License Agreements.
As of December 31, 2025 and 2024, the Company owned shares of Surface common stock. Adrienne Graves and Perry J. Sternberg, directors of the Company, also are directors of Surface. Mark L. Baum, who is the Company’s Chief Executive Officer, was previously a member of the Surface board of directors and resigned from his position as a director of Surface on March 31, 2023.
| F-26 |
NOTE 7. INVENTORIES
Inventories are comprised of finished compounded formulations, over-the-counter and prescription retail pharmacy products, branded pharmaceutical products, including those held at the Company’s 3PL partners, related laboratory supplies and APIs. The composition of inventories as of December 31, 2025 and 2024 was as follows:
| 2025 | 2024 | |||||||
| Raw materials | $ | $ | ||||||
| Work in progress | ||||||||
| Finished goods | ||||||||
| Total inventories | $ | $ | ||||||
NOTE 8. PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses and other current assets at December 31, 2025 and 2024 consisted of the following:
| 2025 | 2024 | |||||||
| Prepaid insurance | $ | $ | ||||||
| Prepaid computer software related expenses | ||||||||
| Prefunded Co-Pay assistance | ||||||||
| Other prepaid expenses | ||||||||
| Receivable due from Melt | ||||||||
| Annual user fees (PDUFA) | ||||||||
| Deposits and other current assets | ||||||||
| Total prepaid expenses and other current assets | $ | $ | ||||||
NOTE 9. PROPERTY, PLANT AND EQUIPMENT
Property, plant and equipment, net at December 31, 2025 and 2024 consisted of the following:
| 2025 | 2024 | |||||||
| Computer hardware | $ | $ | ||||||
| Furniture and equipment | ||||||||
| Lab and pharmacy equipment | ||||||||
| Leasehold improvements | ||||||||
| Accumulated depreciation | ( | ) | ( | ) | ||||
| Total property, plant and equipment, net | $ | $ | ||||||
The
Company recorded depreciation and amortization expense of $
| F-27 |
NOTE 10. CAPITALIZED SOFTWARE COSTS
Capitalized software costs at December 31, 2025 and 2024 consisted of the following:
| 2025 | 2024 | |||||||
| Capitalized internal-use software development costs | $ | $ | ||||||
| Acquired third-party software license for internal use | ||||||||
| Total gross capitalized software for internal use | ||||||||
| Accumulated amortization | ( | ) | ( | ) | ||||
| $ | $ | |||||||
The
Company recorded amortization expense of $
NOTE 11. INTANGIBLE ASSETS AND GOODWILL
The Company’s intangible assets at December 31, 2025 consisted of the following:
| Weighted-average | Accumulated | Net | ||||||||||||
| useful life (in years) | Cost | amortization | Carrying value | |||||||||||
| Patents | $ | $ | ( | ) | $ | |||||||||
| Licenses | ( | ) | ||||||||||||
| Trademarks | ||||||||||||||
| Acquired product rights | ( | ) | ||||||||||||
| Customer relationships | ( | ) | ||||||||||||
| Trade name | ( | ) | ||||||||||||
| State pharmacy licenses | ( | ) | ||||||||||||
| $ | $ | ( | ) | $ | ||||||||||
The Company’s intangible assets at December 31, 2024 consisted of the following:
| Weighted-average | Net | |||||||||||||||||
| useful life | Accumulated | Carrying | ||||||||||||||||
| (in years) | Cost | amortization | Impairment | value | ||||||||||||||
| Patents | $ | $ | ( | ) | $ | ( | ) | $ | ||||||||||
| Licenses | ( | ) | ||||||||||||||||
| Trademarks | ||||||||||||||||||
| Acquired product rights | ( | ) | ||||||||||||||||
| Customer relationships | ( | ) | ||||||||||||||||
| Trade name | ( | ) | ||||||||||||||||
| Non-competition clause | ( | ) | ||||||||||||||||
| State pharmacy licenses | ( | ) | ||||||||||||||||
| $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||
During
the years ended December 31, 2025, 2024 and 2023, the Company recorded a charge of $
| F-28 |
Amortization expense for intangible assets for the years ended December 31, 2025, 2024 and 2023 were as follows:
| For the Years Ended December 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Patents | $ | $ | $ | |||||||||
| Licenses | ||||||||||||
| Acquired product rights | ||||||||||||
| Customer relationships | ||||||||||||
| Trade name | ||||||||||||
| $ | $ | $ | ||||||||||
Estimated future amortization expense for the Company’s intangible assets at December 31, 2025 is as follows:
| Years ending December 31, | ||||
| 2026 | $ | |||
| 2027 | ||||
| 2028 | ||||
| 2029 | ||||
| 2030 | ||||
| Thereafter | ||||
| $ | ||||
In
connection with the license agreement with Novaliq, the Company recognized a commercial milestone of $
There
were
NOTE 12. ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses at December 31, 2025 and 2024 consisted of the following:
| 2025 | 2024 | |||||||
| Accounts payable | $ | $ | ||||||
| Accrued interest | ||||||||
| Other | ||||||||
| Total accounts payable and accrued expenses | $ | $ | ||||||
NOTE 13. DEBT
Fifth Third Revolving Credit Facility - Undrawn
In
September 2025, the Company entered into a Credit Agreement (the “5/3 Revolver”) with Fifth Third Bank, National Association
as administrative agent for itself and the other lenders, providing for a senior secured revolving credit facility in the initial principal
amount of $
| F-29 |
Borrowings
under the 5/3 Revolver bear interest at a floating rate equal to, at the Company’s option, either (i) a base rate plus a margin
ranging from
Under the 5/3 Revolver, the Company is subject to certain customary affirmative and negative covenants. In addition, the 5/3 Revolver contains certain financial covenants requiring the Company to maintain, on a consolidated basis as of the last day of each month, a fixed charge coverage ratio of at least 1.10 to 1.0.
Deferred
financing costs of $
As of December 31, 2025, there were no borrowings drawn or outstanding under the 5/3 Revolver. The Company was in compliance with all covenants and the entire initial principal amount is available as of December 31, 2025.
8.625% Senior Notes Due 2030
In
September 2025, the Company closed a private offering of $
The
2030 Notes are senior unsecured obligations of the Company. The 2030 Notes are effectively subordinated to any of the Company’s
secured indebtedness to the extent of the value of the assets securing such indebtedness. The 2030 Notes are guaranteed on a senior unsecured
basis by the Company, subject to certain exceptions. The 2030 Notes bear interest at the rate of
The
Company may redeem all or part of the 2030 Notes prior to September 15, 2027, at a price equal to 100% of the principal amount of the
2030 Notes redeemed, plus accrued and unpaid interest, if any, to, but not including, the redemption date, plus a “make-whole”
premium, as described in the indenture. The Company may redeem all or part of the 2030 Notes on or after September 15, 2027 at the applicable
redemption prices described in the indenture. The Company may also redeem up to 40% of the aggregate principal amount of the 2030 Notes
at any time prior to September 15, 2027, at a redemption price equal to
Interest
expense related to the 2030 Notes totaled $
The terms of our 2030 Notes contain customary affirmative and negative covenants. As of December 31, 2025, the Company was in compliance with all such covenants.
| F-30 |
Oaktree Loan Due 2026 – Paid in Full
In
March 2023, the Company entered into a Credit Agreement and Guaranty (the “Oaktree Loan”) with Oaktree Fund Administration,
LLC, as administrative agent for the lenders (together, “Oaktree”). Including through subsequent amendments to the Oaktree
Loan, the Company had drawn down a total principal loan amount of $
In
September 2025, the Company repaid the $
Interest
expense related to the Oaktree Loan totaled $
HROWL – 8.625% Senior Notes Due 2026 – Paid in Full
In
April 2021, the Company closed an offering of $
In
September 2025, the Company fully repaid the principal balance of $
Interest
expense related to the 2026 Notes totaled $
HROWM - 11.875% Senior Notes Due 2027 – Paid in Full
In
December 2022 and in January 2023, the Company closed an offering of $
In
September 2025, the Company fully repaid the principal balance of $
| F-31 |
Interest
expense related to the 2027 Notes totaled $
B. Riley Loan and Security Agreement – Paid in Full
On
December 14, 2022 (the “Effective Date”), the Company entered into a Loan and Security Agreement (the “BR Loan”)
with B. Riley Commercial Capital, LLC, as administrative agent for the lenders. The BR Loan provided for a loan facility of up to $
In
January 2023, $
Interest
expense related to the BR Loan totaled $
The
Company recognized a loss on extinguishment of debt in 2025 of $
A summary of the Company’s debt at December 31, 2025 and 2024 is described as follows:
| 2025 | 2024 | |||||||
| $ | $ | |||||||
| Oaktree Loan due | ||||||||
| Less: Unamortized debt issuance costs | ( | ) | ( | ) | ||||
| $ | $ | |||||||
The
total effective interest rate of the Company’s debt was
At December 31, 2025, future minimum payments under the Company’s debt were as follows:
| Amount | ||||
| 2026 | $ | |||
| 2027 | ||||
| 2028 | ||||
| 2029 | ||||
| 2030 | ||||
| Thereafter | ||||
| Total minimum payments | ||||
| Less: unamortized discount, net of premium | ( | ) | ||
| Notes payable, net of unamortized discount | $ | |||
| F-32 |
NOTE 14. LEASES
The
Company leases office and laboratory space under the non-cancelable operating leases listed below. These lease agreements have
remaining terms between one to
| ● | An operating lease for
|
| ● | An operating lease for
|
| ● | An operating lease for
|
| ● | An operating lease for
|
At
December 31, 2025 and 2024, the weighted-average discount rate and the weighted-average remaining lease term for the operating leases
held by the Company were
During
the years ended December 31, 2025, 2024 and 2023, cash paid for amounts included for the operating lease liabilities were $
Future lease payments under operating leases as of December 31, 2025 were as follows:
| Year Ending December 31, | ||||
| 2026 | $ | |||
| 2027 | ||||
| 2028 | ||||
| 2029 | ||||
| 2030 | ||||
| Thereafter | ||||
| Total minimum lease payments | ||||
| Less: amount representing interest payments | ( | ) | ||
| Total operating lease liabilities | ||||
| Less: current portion, operating lease liabilities | ( | ) | ||
| Operating lease liabilities, net of current portion | $ | |||
Preferred Stock
At December 31, 2025 and 2024, the Company had shares of preferred stock, $ par value, authorized and shares of preferred stock issued and outstanding.
| F-33 |
Common Stock
The
Company primarily issues common stock through the exercise of stock options and the vesting of performance stock awards and restricted
stock awards, and allows participants to net-share settle the acquisition price and tax withholding liabilities. During the years ended
December 31, 2025, 2024 and 2023, the net shares issued in satisfaction of these share-based compensation awards was ,
and , respectively. During the year ended December 31, 2023, the Company closed a public offering of shares of its common stock
at an offering price of $ per share (the “Offering”). The Company sold shares of its common stock in the Offering,
resulting in the Company receiving aggregate net proceeds of $
Stock Option Plan
On September 17, 2007, the Company’s Board of Directors and stockholders adopted the Company’s 2007 Incentive Stock and Awards Plan, as subsequently amended (the “2007 Plan:). The 2007 Plan reached its term in September 2017, and the Company can no longer issue additional awards under this plan, however, options previously issued under the 2007 Plan will remain outstanding until they are exercised, reach their maturity or are otherwise cancelled/forfeited. On June 13, 2017, the Company’s Board of Directors and stockholders adopted the Company’s 2017 Incentive Stock and Awards Plan which was subsequently amended on June 3, 2021 (as amended, the “2017 Plan”). The 2017 Plan provides for the issuance of a maximum of shares of the Company’s common stock and expires in June 2027. In connection with the approval of the 2025 Plan (as described below), the Company can no longer issue additional awards under the 2017 Plan, however, equity awards previously issued under the 2017 Plan will remain outstanding pursuant to their terms. On June 18, 2025, the Company’s stockholders adopted the Company’s 2025 Incentive Stock and Awards Plan (the “2025 Plan” and together with the 2007 Plan and 2017 Plan, the “Plans”). As of December 31, 2025, the 2025 Plan provides for the issuance of a maximum of shares of the Company’s common stock. The purpose of the Plans is to attract and retain directors, officers, consultants, advisors and employees whose services are considered valuable, to encourage a sense of proprietorship and to stimulate an active interest of such persons in the Company’s development and financial success. Under the Plans, the Company is authorized to issue incentive stock options intended to qualify under Section 422 of the Internal Revenue Code of 1986, as amended, non-qualified stock options, restricted stock units and restricted stock. The Plans are administered by the Compensation Committee of the Company’s Board of Directors. The Company had no shares available for future issuances under the 2017 Plan and shares available for future issuance under the 2025 plan as of December 31, 2025.
Stock Options
| Number of shares | Weighted Avg. Exercise Price | Weighted Avg. Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||||
| Options outstanding – January 1, 2025 | $ | |||||||||||||||
| Options granted | $ | |||||||||||||||
| Options exercised | ( | ) | $ | |||||||||||||
| Options cancelled/forfeited | ( | ) | $ | |||||||||||||
| Options outstanding – December 31, 2025 | $ | $ | ||||||||||||||
| Options exercisable | $ | $ | ||||||||||||||
| Options vested and expected to vest | $ | $ | ||||||||||||||
The aggregate intrinsic value in the tables above represents the total pre-tax amount of the proceeds, net of exercise price, which would have been received by option holders if all option holders had exercised and immediately sold all options with an exercise price lower than the market price on December 31, 2025, based on the closing price of the Company’s common stock of $ on that date.
| F-34 |
The intrinsic value of the options exercised in 2025, 2024 and 2023 was $, $ and $, respectively. During 2025, 2024 and 2023, the Company recognized tax benefit from stock options exercised during these periods.
During the year ended December 31, 2025, the Company granted stock options to certain employees. . Certain option awards provide for accelerated vesting if there is a change in control (as defined in the Plans) and in the event of certain modifications to the option award agreement.
The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton option pricing model. The Company calculates expected volatility based solely on the historical volatilities of the common stock of the Company. The expected term of options granted was determined in accordance with the “simplified approach,” as the Company has limited, relevant, historical data on employee exercises and post-vesting employment termination behavior. The expected risk-free interest rate is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. The financial statement effect of forfeitures is estimated at the time of grant and revised, if necessary, if the actual effect differs from those estimates. For option grants to employees and directors, the Company assigns a forfeiture factor of %. These factors could change in the future, which would affect the determination of stock-based compensation expense in future periods. Utilizing these assumptions, the fair value is determined at the date of grant.
| 2025 | 2024 | 2023 | ||||||||||
| Weighted-average fair value of options granted | $ | $ | $ | |||||||||
| Expected terms (in years) | ||||||||||||
| Expected volatility | % | – | % | – | % | |||||||
| Risk-free interest rate | – | % | – | % | – | % | ||||||
| Dividend yield | ||||||||||||
| Options Outstanding | Options Exercisable | |||||||||||||||||||||
| Range of Exercise Price | Number Outstanding | Weighted Average Remaining Contractual Life in Years | Weighted Average Exercise Price | Number Exercisable | Weighted Average Exercise Price | |||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
As of December 31, 2025, there was approximately $ of total unrecognized compensation expense related to unvested stock options granted under the Plan. That expense is expected to be recognized over the weighted-average remaining vesting period of years. The stock-based compensation for all stock options was $, $ and $ during the years ended December 31, 2025, 2024 and 2023, respectively.
| F-35 |
Performance Stock Units
Grants During the Year Ended December 31, 2025
In July 2025, the Company granted an aggregate of performance stock units to Mark L. Baum, Chief Executive Officer, and Andrew R. Boll, President and Chief Financial Officer, which are subject to the satisfaction of certain market-based and continued service conditions (the “2025 PSUs”). The vesting of the 2025 PSUs require (i) a minimum of a three-year service period and (ii) during a five-year term, the achievement and maintenance of Company common stock price targets ranging between $ to $ per share, broken out into four separate tranches as described further in the table below.
| Tranche | Target Stock Price | Number of Shares | ||||||
| Tranche 1 | $ | |||||||
| Tranche 2 | $ | |||||||
| Tranche 3 | $ | |||||||
| Tranche 4 | $ | |||||||
| * | Target Stock Price assumes that no dividends or like distributions are made to stockholders of the Company. If such distributions are made, the Target Stock Price would decrease accordingly, to the benefit of the employee, to account for the dividend/distribution as a part of the Target Stock Price. |
The aggregate fair value of the 2025 PSUs was $ using a Monte Carlo Simulation with a life, volatility and a risk-free interest rate of . This amount is being amortized over a derived service period.
Grants During the Year Ended December 31, 2023
In April 2023, the Company granted an aggregate of PSUs to members of its senior management including Mark Baum, Chief Executive Officer, Andrew Boll, Chief Financial Officer, and John Saharek, Chief Commercial Officer, which are subject to the satisfaction of certain market-based and continued service conditions (the “2023 PSUs”). The vesting of the 2023 PSUs required (i) a minimum of a two-year service period and (ii) during a five-year term, the achievement and maintenance of Company common stock price targets for ten consecutive trading days ranging between $ to $ per share. In 2025, all of the vesting conditions were met and the Company issued all of the shares, less withheld to satisfy minimum statutory withholding taxes.
The aggregate fair value of the 2023 PSUs was $ using a Monte Carlo Simulation with a life, volatility and a risk-free interest rate of . This amount is being amortized over a derived service period.
| Number of PSUs | Weighted Average Grant Date Fair Value | |||||||
| PSUs unvested – January 1, 2025 | $ | |||||||
| PSUs granted | $ | |||||||
| PSUs vested | ( | ) | $ | |||||
| PSUs cancelled/forfeited | $ | |||||||
| PSUs unvested – December 31, 2025 | $ | |||||||
| F-36 |
As of December 31, 2025, the total unrecognized compensation expense related to unvested PSUs was approximately $ which is expected to be recognized over years, based on estimated vesting schedules. The stock-based compensation for PSUs was $, $ and $ during the years ended December 31, 2025, 2024 and 2023, respectively. During 2025, 2024 and 2023, the Company recognized tax benefit from the vesting of PSUs during these periods.
Restricted Stock Units
RSU awards are granted subject to certain vesting requirements and other restrictions, including performance and market-based vesting criteria. The grant date fair value of the RSUs, which has been determined based upon the market value of the Company’s common stock on the grant date, is expensed over the vesting period of the RSUs.
Grants During the Year Ended December 31, 2025
During the year ended December 31, 2025, the Company’s non-employee members of the Board of Directors were granted time-based vesting RSUs with a fair market value of $, which vest over one year. The Company also granted time-based vesting RSUs with a fair market value of $ to certain employees and consultants. Vesting terms for RSUs granted to employees and consultants during the year ended December 31, 2025 generally vest in equal installments over or and vest in equal quarterly installments over one year.
Grants During the Year Ended December 31, 2024
During the year ended December 31, 2024, the Company’s non-employee members of the Board of Directors were granted time-based vesting RSUs with a fair market value of $, which vest in equal quarterly installments over one year. The Company also granted time-based vesting RSUs with a fair market value of $ to certain employees and consultants. Vesting terms for RSUs granted to employees and consultants during the year ended December 31, 2024 generally vest in equal installments over or and vest in equal quarterly installments over one year.
Grants During the Year Ended December 31, 2023
During the year ended December 31, 2023, the Company’s non-employee members of the Board of Directors were granted time-based vesting RSUs with a fair market value of $, which vest in equal quarterly installments over one year. The Company also granted time-based vesting RSUs with a fair market value of $ to certain employees, which vest in full on the anniversary of the grant date.
| Number of RSUs | Weighted Average Grant Date Fair Value | |||||||
| RSUs unvested – January 1, 2025 | $ | |||||||
| RSUs granted | $ | |||||||
| RSUs vested | ( | ) | $ | |||||
| RSUs cancelled/forfeited | ( | ) | $ | |||||
| RSUs unvested at December 31, 2025 | $ | |||||||
| F-37 |
As
of December 31, 2025, the total unrecognized compensation expense related to unvested RSUs was approximately $ which is expected
to be recognized over a weighted-average period of years, based on estimated vesting schedules. The stock-based compensation for
RSUs was $, $ and $ during the years ended December 31, 2025, 2024 and 2023, respectively. During 2025, 2024
and 2023, the Company recognized a tax benefit of $
| For the Years Ended December 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Employees - selling, general and administrative | $ | $ | $ | |||||||||
| Employees - R&D | ||||||||||||
| Directors - selling, general and administrative | ||||||||||||
| Consultants - selling, general and administrative | ||||||||||||
| Total | $ | $ | $ | |||||||||
NOTE 16. INCOME TAXES
The Company is subject to taxation in the U.S., New Jersey, Tennessee, and various other states. All of the Company’s pre-tax income is generated in the U.S. The Company’s income tax provision consists of the following for the year ended December 31, 2025:
| 2025 | ||||
| Current | ||||
| Federal | $ | |||
| State and Local | ||||
| Foreign | ||||
| Total current | ||||
| Deferred | ||||
| Federal | ||||
| Foreign | ||||
| State and Local | ||||
| Total deferred | ||||
| Income Tax Provision | $ | |||
A reconciliation of income taxes computed by applying the statutory U.S. income tax rate to the Company’s loss before income taxes to the income tax provision for the year ended December 31, 2025 is as follows:
| Year Ended December 31, 2025 | ||||||||
| Amount | Percent | |||||||
| U.S. Federal Statutory Tax Rate | $ | ( | ) | % | ||||
| State and Local Income Taxes, Net of Federal Income Tax Effect(1) | ( | ) | ||||||
| Tax Credits | ||||||||
| Research and Development Credits | ( | ) | ||||||
| Changes in Valuation Allowances | ( | ) | ||||||
| Nontaxable or Nondeductible Items | ||||||||
| Executive Compensation Limitations | ( | ) | ||||||
| Stock Compensation Windfalls | ( | ) | ||||||
| Transaction Fees | ( | ) | ||||||
| Incentive Stock Options | ( | ) | ||||||
| Meals and Entertainment | ( | ) | ||||||
| Other Permanent Adjustments | ( | ) | ||||||
| Changes in Unrecognized Tax Benefits | ( | ) | ||||||
| Other Adjustments | ||||||||
| Return to Provision | ( | ) | ||||||
| Effective Income Tax Rate | $ | ( | )% | |||||
| (1) |
The Company’s income tax provision consists of the following for the years ended December 31, 2024 and 2023:
| December 31, | ||||||||
| 2024 | 2023 | |||||||
| Current: | ||||||||
| Federal | $ | $ | ||||||
| State | ||||||||
| Total current | $ | $ | ||||||
| Deferred: | ||||||||
| Federal | $ | $ | ||||||
| State | ||||||||
| Total deferred | ||||||||
| Income tax provision | $ | $ | ||||||
| F-38 |
A reconciliation of income taxes computed by applying the statutory U.S. income tax rate to the Company’s loss before income tax provision to the income tax provision for the years ended December 31, 2024 and 2023 is as follows:
| December 31, | ||||||||
| 2024 | 2023 | |||||||
| U.S. federal statutory tax rate | % | % | ||||||
| State tax benefit, net | ( | )% | % | |||||
| Rate change | % | ( | )% | |||||
| Employee stock-based compensation | ( | )% | % | |||||
| Excess Employee remuneration | ( | )% | ( | )% | ||||
| Melt loan settlement | % | ( | )% | |||||
| Other | % | % | ||||||
| Uncertain tax positions | % | ( | )% | |||||
| Research and development tax credit | % | % | ||||||
| Provision-to-return true-ups | ( | )% | % | |||||
| Other true-ups | ( | )% | ( | )% | ||||
| % | ( | )% | ||||||
| Change in valuation allowance | ( | )% | % | |||||
| Effective income tax rate | ( | )% | ( | )% | ||||
Deferred tax assets and liabilities reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets as of December 31, 2025 and 2024 are as follows:
| 2025 | 2024 | |||||||
| Deferred tax assets | ||||||||
| Net operating loss carryforwards | $ | $ | ||||||
| Interest expense limitation carryforwards | ||||||||
| Capitalized research & experimentation | ||||||||
| Amortization | ||||||||
| Accrued chargebacks & returns | ||||||||
| Stock compensation | ||||||||
| Investment in Melt Pharmaceuticals | ||||||||
| Accrued rebates | ||||||||
| Lease liability | ||||||||
| Accrued bonus | ||||||||
| Obsolete inventory reserve | ||||||||
| Accrued vacation | ||||||||
| Research credit carryforwards | ||||||||
| Other accruals and reserves | ||||||||
| Total deferred tax assets | ||||||||
| Valuation allowances | ( | ) | ( | ) | ||||
| Net deferred tax assets | ||||||||
| Deferred tax liabilities | ||||||||
| Prepaid expenses | ||||||||
| Tax accounting method change | ||||||||
| Depreciation | ||||||||
| Right-of-use assets | ||||||||
| Total deferred tax liabilities | ||||||||
| Net deferred tax liability | $ | $ | ||||||
As
of December 31, 2025, the Company had federal net operating loss carryforwards of approximately $
Realization of deferred tax assets is dependent upon future taxable income, if any, the timing and amount of which are uncertain. Accordingly, a valuation allowance has been recorded for the amount of the net deferred tax assets. A rollforward of the valuation allowance is as follows:
| 2025 | 2024 | |||||||
| Beginning balance | $ | $ | ||||||
| Increases to the valuation allowance | ||||||||
| Decreases to the valuation allowance | ||||||||
| Ending balance | $ | $ | ||||||
During
the year ended December 31, 2025, the Company did not pay any Federal income tax and paid $
| Tennessee | $ | |||
| Colorado | ||||
| New Jersey | ||||
| Massachusetts | ||||
| Other | ||||
| $ |
| F-39 |
As
of December 31, 2025 and 2024, the Company had approximately $
A reconciliation of the change in the unrecognized tax benefits balance for the years ended December 31, 2025 and 2024 is as follows:
| 2025 | 2024 | |||||||
| Beginning balance | $ | $ | ||||||
| Additions for tax positions related to current year | ||||||||
| Additions (reductions) for tax positions related to prior years | ( | ) | ||||||
| Reductions to tax positions related to lapse of statute | ( | ) | ||||||
| Ending balance | $ | $ | ||||||
NOTE 17. EMPLOYEE SAVINGS PLAN
The
Company has established an employee savings plan pursuant to Section 401(k) of the Internal Revenue Code, effective January 1, 2014.
The plan allows participating employees to deposit into tax deferred investment accounts up to
NOTE 18. COMMITMENTS AND CONTINGENCIES
Legal
General and Other
In the ordinary course of business, the Company is involved in various legal proceedings, government investigations and other matters that are complex in nature and have outcomes that are difficult to predict. See also Part I, Item 1A. Risk Factors. The Company describes legal proceedings and other matters that are/were significant or that it believes could become significant in this footnote.
The Company records accruals for loss contingencies to the extent that it concludes it is probable that a liability has been incurred and the amount of the related loss can be reasonably estimated. The Company evaluates, on a quarterly basis, developments in legal proceedings and other matters that could cause an increase or decrease in the amount of a liability that has been accrued previously.
The Company’s legal proceedings involve various aspects of its business and a variety of claims, some of which present novel factual allegations and/or unique legal theories. Typically, a number of the matters pending against the Company are at early stages of the legal process, which in complex proceedings of the sort the Company face often extend for several years. While it is not possible to accurately predict or determine the eventual outcomes of matters that have not concluded, an adverse determination in one or more of these matters (whether discussed in this footnote or not) currently pending may have a material adverse effect on the Company’s consolidated results of operations, financial position or cash flows. Legal costs incurred for loss contingencies are expensed as incurred.
| F-40 |
Certain recent developments concerning the Company’s legal proceedings it believes are or were material to its business and other matters are discussed below:
Ocular Science, Inc. et. al
In
July 2021, ImprimisRx, LLC, a subsidiary of the Company, filed a lawsuit against Ocular Science, Inc. and OSRX, Inc. (together, “OSRX”)
in the U.S. District Court for the Southern District of California, asserting claims for copyright infringement, trademark infringement,
unfair competition and false advertising (Lanham Act). Since July 2021, the complaint had been amended and OSRX added counterclaims alleging
ImprimisRx, LLC was violating the Lanham Act with false advertising. The Court granted cross motions for summary judgment on each party’s
Lanham Act claims, thus leaving only ImprimisRx, LLC’s copyright infringement, trademark infringement, and unfair competition claims
for trial. Following a jury trial in November 2024, a jury found OSRX acted with malice, fraud, or oppression, willfully engaging in
trademark infringement and unfair competition under California and federal law, and ImprimisRx, LLC received a $
Product and Professional Liability
Product and professional liability litigation represents an inherent risk to all firms in the pharmaceutical and pharmacy industry. The Company utilizes traditional third-party insurance policies with regard to our product and professional liability claims. Such insurance coverage at any given time reflects current market conditions, including cost and availability, when the policy is written.
Indemnities
In addition to the indemnification provisions contained in the Company’s charter documents, the Company generally enters into separate indemnification agreements with each of the Company’s directors and officers. These agreements require the Company, among other things, to indemnify the director or officer against specified expenses and liabilities, such as attorneys’ fees, judgments, fines and settlements, paid by the individual in connection with any action, suit or proceeding arising out of the individual’s status or service as the Company’s director or officer, other than liabilities arising from willful misconduct or conduct that is knowingly fraudulent or deliberately dishonest, and to advance expenses incurred by the individual in connection with any proceeding against the individual with respect to which the individual may be entitled to indemnification by the Company. Several of the Company’s asset purchase and license agreements contain customary representations, warranties, covenants and confidentiality provisions, and also contain mutual indemnification obligations related primarily to performance under the respective agreements. The Company also indemnifies its lessors in connection with its facility leases for certain claims arising from the use of the facilities. These indemnities do not provide for any limitation of the maximum potential future payments the Company could be obligated to make. Historically, the Company has not incurred any payments for these obligations and, therefore, no liabilities have been recorded for these indemnities in the accompanying consolidated balance sheets.
Asset Purchase, License and Related Agreements
FDA Approved Product Acquisitions
In recent years, the Company has acquired commercial and product rights to various FDA approved ophthalmic medications and products through asset purchase, licenses, supply and/or other related agreements. In general, in exchange for product and commercial rights these agreements provide the counterparties with certain upfront and contingent milestone payments typically related to certain annual sales amounts and manufacturing events, and in certain cases, per unit transfer prices and royalties on sales of some of the products.
| F-41 |
During
the years ended December 31, 2025, 2024 and 2023, $
Contract Manufacturing
The
Company has entered into manufacturing agreements with respect to third-party contract manufacturers for its FDA approved pharmaceutical
products. Some of these contract manufacturing agreements require minimum annual order amounts. The Company has committed to pay approximately
$
NOTE 19. SEGMENTS AND CONCENTRATIONS
The
chief operating decision maker (“CODM”) is the Chief Executive Officer. The CODM does not review segment assets when assessing
segment performance and deciding how to allocate resources. The Company reports on
| ● | The Branded segment includes activities of the Company’s FDA approved ophthalmology pharmaceutical products, including the out-licensing of rights to certain of our branded products. | |
| ● | The ImprimisRx segment represents activities in the Company’s ophthalmology-focused pharmaceutical compounding business. |
The CODM evaluates segment performance and makes resource-allocation decisions primarily on the basis of segment contribution. Segment contribution is the internal measure of profitability that the CODM reviews on a regular basis to assess the operational performance of each segment, determine the appropriate level of sales and marketing investments, evaluate pricing decisions, and prioritize capital deployment among branded product initiatives and the ImprimisRx compounding operations.
Segment contribution for the segments represents net revenues less cost of sales, certain general and administrative expenses, selling and marketing expenses, and research and development expenses. The Company does not evaluate the following items at the segment level:
| ● | Selling, general and administrative expenses that result from shared infrastructure, including certain expenses associated with legal matters, public company costs (e.g. investor relations), Board of Directors and principal executive officers and other like shared expenses. | |
| ● | Operating expenses within selling, general and administrative expenses that result from the impact of corporate initiatives. Corporate initiatives primarily include integration, restructuring, acquisition and other shared costs. | |
| ● | Other select revenues and operating expenses that are not segment specific including research and development expenses, amortization, and asset sales and impairments, net as not all such information has been accounted for at the segment level, or such information has not been used by all segments. |
| F-42 |
Segment net revenues, segment operating expenses and segment contribution information consisted of the following:
| Year Ended December 31, 2025 | ||||||||||||
| Branded | ImprimisRx | Consolidated | ||||||||||
| Product sales, net | $ | $ | $ | |||||||||
| Other revenues | ||||||||||||
| Total revenues | ||||||||||||
| Cost of sales | ( | ) | ( | ) | ( | ) | ||||||
| Gross profit | ||||||||||||
| Operating expenses: | ||||||||||||
| Selling, general and administrative | ||||||||||||
| Research and development | ||||||||||||
| Segment contribution | $ | $ | ||||||||||
| Corporate | ||||||||||||
| Research and development | ||||||||||||
| Income from operations | $ | |||||||||||
| Year Ended December 31, 2024 | ||||||||||||
| Branded | ImprimisRx | Consolidated | ||||||||||
| Product sales, net | $ | $ | $ | |||||||||
| Other revenues | ||||||||||||
| Total revenues | ||||||||||||
| Cost of sales | ( | ) | ( | ) | ( | ) | ||||||
| Gross profit | ||||||||||||
| Operating expenses: | ||||||||||||
| Selling, general and administrative | ||||||||||||
| Research and development | ||||||||||||
| Segment contribution | $ | $ | ||||||||||
| Corporate | ||||||||||||
| Research and development | ||||||||||||
| Income from operations | $ | |||||||||||
| Year Ended December 31, 2023 | ||||||||||||
| Branded | ImprimisRx | Consolidated | ||||||||||
| Product sales, net | $ | $ | $ | |||||||||
| Other revenues | ||||||||||||
| Total revenues | ||||||||||||
| Cost of sales | ( | ) | ( | ) | ( | ) | ||||||
| Gross profit | ||||||||||||
| Operating expenses: | ||||||||||||
| Selling, general and administrative | ||||||||||||
| Research and development | ||||||||||||
| Segment contribution | $ | $ | ||||||||||
| Corporate | ||||||||||||
| Research and development | ||||||||||||
| Income from operations | $ | |||||||||||
Substantially all revenue is attributable to the U.S. All long-lived assets at December 31, 2025 and 2024 were located in the U.S.
| F-43 |
Revenues by segment are further described as follows:
| For the Years Ended December 31, | ||||||||||||||||||||||||
| 2025 | % | 2024 | % | 2023 | % | |||||||||||||||||||
| IHEEZO | $ | % | $ | % | $ | % | ||||||||||||||||||
| VEVYE | % | % | % | |||||||||||||||||||||
| Other branded products | % | % | % | |||||||||||||||||||||
| Other revenues, net | % | % | % | |||||||||||||||||||||
| Branded revenue, net | % | % | % | |||||||||||||||||||||
| ImprimisRx revenue, net | % | % | % | |||||||||||||||||||||
| Total revenues, net. | $ | % | $ | % | $ | % | ||||||||||||||||||
Other than IHEEZO, VEVYE, and one ImprimisRx product, no other products accounted for more than 10% of total revenues for the periods presented.
Customer and Supplier Concentrations
Substantially
all of the Company’s Branded sales are made to third-party distributors who sell the products to pharmacies and to the end-users.
There were two customers who comprised more than
The
Company received its manufacturing supplies from three main suppliers during the years ended December 31, 2025 and 2023 and two main
suppliers during the year ended December 31, 2024. These suppliers collectively accounted for
NOTE 20. SUBSEQUENT EVENTS
In
January 2026, the Company and Santen agreed to amend the Santen Agreements. Pursuant to the amendment, the parties agreed to a full
and final settlement of all contingent milestone obligations related to specified manufacturing-related events for the Santen
Products in exchange for a one-time lump sum payment by the Company of $
Subsequent to December 31, 2025, options to purchase shares of common stock were exercised at a weighted-average exercise price of $ per share.
| F-44 |
EXHIBIT 4.1
DESCRIPTION OF THE COMPANY’S SECURITIES
REGISTERED PURSUANT TO SECTION 12 OF THE SECURITIES
EXCHANGE ACT OF 1934
Harrow, Inc. has one class of securities registered under Section 12 of the Securities Exchange Act of 1934, as amended: our common stock, par value $0.001 per share.
In this exhibit, when we refer to “Company”, “Harrow”, “we”, “us” and “our” or when we otherwise refer to ourselves, we mean Harrow, Inc., excluding, unless otherwise expressly stated or the context requires, our subsidiaries.
Description of Capital Stock
The following is a summary of the rights of our common and preferred stock and of certain provisions of our Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws. For more detailed information, please see our Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws, which are incorporated by reference as exhibits to the Annual Report on Form 10-K to which this description is an exhibit.
Authorized Capital Stock
Our authorized capital stock consists of 55,000,000 shares, 50,000,000 of which are designated as common stock, par value $0.001 per share, and 5,000,000 of which are designated as preferred stock, par value $0.001 per share. As of February 25, 2026 there were 37,229,705 shares of our common stock and no shares of our preferred stock issued and outstanding.
Capital Stock Issued and Outstanding
As of February 25, 2026, there were approximately 56 stockholders of record (excluding an indeterminable number of stockholders whose shares are held in street or “nominee” name) of our common stock. In addition, as of December 31, 2025, there are outstanding (i) options to acquire 1,637,072 shares of our common stock with a weighted average exercise price of $9.86 per share, (ii) 1,509,462 unvested restricted and performance-based stock units and (iii) 216,483 restricted stock units award to directors that had vested, but issuance and delivery of the shares are deferred until the director resigns or otherwise leaves the Board of Directors.
Description of Common Stock
We are authorized to issue 50,000,000 shares of common stock, par value $0.001 per share. The holders of our common stock are entitled to one vote per share on all matters submitted to a vote of the stockholders, including the election of directors. Our Amended and Restated Certificate of Incorporation does not provide for cumulative voting in the election of directors. Subject to any preferential rights of any outstanding series of preferred stock created by our Board of Directors from time to time the holders of our common stock will be entitled to cash dividends as may be declared, if any, by our Board of Directors from funds available. Subject to any preferential rights of any outstanding series of preferred stock that we may issue, upon liquidation, dissolution or winding up of our company, the holders of our common stock will be entitled to receive pro rata all assets available for distribution to the holders.
| 1 |
Description of Preferred Stock
Our Board of Directors has the authority, without further action by our stockholders, to issue up to 5,000,000 shares of preferred stock, par value $0.001 per share, in one or more series. Our Board of Directors may designate the rights, preferences, privileges and restrictions of the preferred stock, including dividend rights, conversion rights, voting rights, terms of redemption, liquidation preference, sinking fund terms, and number of shares constituting any series and the designation of any series. The issuance of preferred stock could have the effect of restricting dividends on our common stock, diluting the voting power of our common stock, impairing the liquidation rights of our common stock, or delaying or preventing a change in control. The ability to issue preferred stock could delay or impede a change in control.
Anti-Takeover Provisions
We are subject to the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, an anti-takeover law. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a “business combination’’ with an “interested stockholder’’ for a period of three years after the date of the transaction in which such stockholder became an interested stockholder, unless the business combination is approved in a prescribed manner. For purposes of Section 203, a “business combination’’ includes a merger, asset sale or other transaction resulting in a financial benefit to the interested stockholder, and an “interested stockholder’’ is a stockholder who, together with affiliates and associates, owns, or within three years prior, did own, 15% or more of the voting stock.
Liability and Indemnification of Directors and Officers
Section 145 of the DGCL provides, in general, that a corporation incorporated under the laws of the State of Delaware, such as us, may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding (other than a derivative action by or in the right of the corporation) by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe such person’s conduct was unlawful. In the case of a derivative action, a Delaware corporation may indemnify any such person against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification will be made in respect of any claim, issue or matter as to which such person will have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery of the State of Delaware or any other court in which such action was brought determines such person is fairly and reasonably entitled to indemnity for such expenses.
Our Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws provide that we will indemnify our directors, officers, employees and agents to the extent and in the manner permitted by the provisions of the DGCL, as amended from time to time, subject to any permissible expansion or limitation of such indemnification, as may be set forth in any stockholders’ or directors’ resolution or by contract.
| 2 |
We also have director and officer indemnification agreements with each of our executive officers and directors that provide, among other things, for the indemnification to the fullest extent permitted or required by Delaware law, provided that such indemnitee shall not be entitled to indemnification in connection with any proceedings or claims initiated or brought voluntarily by the indemnitee and not by way of defense, unless (i) such indemnification is expressly required to be made by law, (ii) the proceeding was authorized by our Board of Directors, (iii) indemnification is provided by us, in our sole discretion, pursuant to powers vested in us under the DGCL, or (iv) the proceeding is brought to establish or enforce a right to indemnification under the indemnification agreement or any other statute or law or otherwise as required under Section 145 of the DGCL. We are not required to indemnify the indemnitee for any amounts paid in settlement of a proceeding unless we consent to such settlement.
Any repeal or modification of these provisions approved by our stockholders shall be prospective only, and shall not adversely affect any limitation on the liability of a director or officer existing as of the time of such repeal or modification.
We have purchased and intend to maintain insurance on our behalf and on behalf of any person who is or was a director or officer against any loss arising from any claim asserted against him or her and incurred by him or her in that capacity, subject to certain exclusions and limits of the amount of coverage.
Listing; Transfer Agent
Our common stock is listed on The NASDAQ Global Market under the symbol “HROW”. The transfer agent and registrar for our common stock is Securities Transfer Corporation, 2901 N. Dallas Parkway, Suite 380, Plano, Texas 75093.
| 3 |
Exhibit 10.15
| 1A Burton Hills Blvd | ||
| Nashville TN 37215 | ||
 |
Main: 858.704.4040 | |
| Fax: 858.345.1745 | ||
| www.harrowinc.com |
October 6, 2025
Dear Frank,
Congratulations on your promotion to President and Chief Executive Officer of ImprimisRx, reporting to Mark L. Baum, Chairman and CEO of Harrow. Your effective date will be October 6, 2025. Your compensation will be $467,999.94. This is a full-time, exempt-level position with an annual discretionary bonus of 40% of your salary earned during the year. While no bonus payment is guaranteed, your potential annual discretionary target bonus will be based on the achievement of certain targets agreed upon by you and your Manager. You must be employed with Harrow on the day of payment to receive a bonus payout.
We hope your new position with Harrow will prove mutually rewarding.
| Sincerely, | |
| /s/ KJ Barratt | |
| KJ Barratt | |
| Chief Talent Officer |
| * | * | * |
| 10/06/2025 | /s/ Frank Mullery | |
| Date | Frank Mullery |
| Page 1 of 1 |
Exhibit 10.21
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
FIRST AMENDMENT TO LICENSE AND SUPPLY AGREEMENT
This First Amendment (the “First Amendment”) is made and entered into as of the 17th day of November, 2022 (the “First Amendment Effective Date”) by and between Sintetica S.A., a Swiss corporation having its principal place of business at Via Penate 5, 6850 Mendrisio, Switzerland, (“Sintetica”), and Harrow IP LLC, a Delaware limited liability company having its principal place of business at 102 Woodmont Blvd., Suite 610, Nashville, TN 37205 USA, (“Harrow”). Sintetica and Harrow are sometimes referred to herein individually as a “Party” and collectively as the “Parties.”
WHEREAS Sintetica and Harrow previously entered into a License and Supply Agreement with a Signing Date of July 25, 2021 (the “Agreement”); and
WHEREAS, Sintetica and Harrow desire to amend the Agreement as set forth herein.
NOW, THEREFORE, for good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, and with the specific intent to be bound hereby, the parties hereby agree as follows:
| 1. | All capitalized terms used in this First Amendment but not defined shall have the meanings ascribed to them under the terms of the Agreement. |
| 2. | Delete Section 1.69 in its entirety. |
| 3. | Replace Section 7.2(a) in its entirety with the following: |
(a) Harrow shall make payments to Sintetica based on achievement of certain milestone events for Product as set forth in the following table:
| Milestone Number | Milestone Event | Milestone Payment (U.S. Dollars) | ||
| (i) | The date on which Sintetica submits the filing to the FDA and pays the PDUFA fee. | [***] | ||
| (ii) | The date the FDA approves the United States NDA for the Product for the Licensed Indication | [***] |
| 4. | Replace Section 7.2(b) in its entirety with the following: |
(b) Sintetica shall notify Harrow upon achievement of each milestone event and Harrow shall pay to Sintetica the amounts as set forth above in Sections 7.2(a)(i) and 7.2(a)(ii)(a) within ten (10) Business Days after the date of achievement of the respective milestone events. Payment of the amounts as set forth above in Sections 7.2(a)(ii)(b) and 7.2(a)(ii)(c) shall be made within sixty (60) days following the end of the applicable calendar year for which such payment is due.
| 5. | Replace Section 7.2(c) in its entirety with the following: |
(c) Notwithstanding the foregoing, the payment of Milestone Numbers 7.2(a)(ii)(a), 7.2(a)(ii)(b) and 7.2(a)(ii)(c) above shall be conditioned on Sintetica’s making available to Harrow sufficient launch quantities of the Product consistent with Harrow’s Commercial Launch Plan for that date.
| Page 1 of 2 |
| 6. | Replace Section 7.2(d) in its entirety with the following: |
(d) The milestone payments for Milestone Numbers 7.2(a)(i), 7.2(a)(ii)(a), 7.2(a)(ii)(b) and 7.2(a)(ii)(c) by Harrow to Sintetica hereunder shall be payable only once and, except as set forth for Milestone Number 7.2(a)(i) in the following Section 7.2(e), shall be non-refundable. Multiple milestone payments can occur in the same calendar year.
| 7. | Delete Section 7.3 in its entirety. |
| 8. | Replace Section 11.7(v) in its entirety with the following: |
(v) Notwithstanding 11.7(iii), for any and all terminations by Sintetica under Section 11.2, or Section 11.3, Section 11.6(a), Section 11.6(d) or Section 11.6(e), all amounts due in accordance with Section 7.2(a)(ii)(b) and Section 7.2(a)(ii)(c) shall become immediately due and payable, as shall the first regulatory milestone set forth in Section 7.2 if Sintetica has already made the PDUFA filing fee payment pursuant to Section 4.2.
| 9. | Replace Section 11.7(vi) in its entirety with the following: |
(vi) Harrow shall have the right to sell or otherwise dispose of any inventory of the Product on hand at the time of such termination, subject to Harrow making any payments to Sintetica in accordance with Section 7.2(a)(ii)(b) and Section 7.2(a)(ii)(c) or Section 7.4.
| 10. | In all other respects, the terms and conditions of the Agreement will remain in full force and effect as written; provided, however, that the terms and conditions of this First Amendment will control over the terms and conditions of the Agreement to the extent there are any inconsistencies between this First Amendment and the Agreement. |
IN WITNESS WHEREOF, the Parties have executed this First Amendment as of the First Amendment Effective Date.
| Sintetica S.A. | ||
| By: | /s/ Nicola Caronzolo | |
| Name: | Nicola Caronzolo | |
| Title: | Corporate CEO | |
| Sintetica S.A. | ||
| By: | /s/ Luca Casella | |
| Name: | Luca Casella | |
| Title: | Corporate CFO | |
| Harrow IP LLC | ||
| By: | /s/ Andrew R. Boll | |
| Name: | Andrew R. Boll | |
| Title: | CFO | |
| Page 2 of 2 |
Exhibit 10.28
Confidential
EXECUTION VERSION
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
License Agreement
dated
jUNE 6, 2023
by
NOVALIQ GMBH
and
HARROW HEALTH, Inc.,
HARROW EYE, LLC and
HARROW IP, LLC
LIST OF EXHIBITS
Schedules and exhibits to this agreement have been omitted pursuant to Item 601(a)(5) of Regulation S-K because they do not contain material information and are not otherwise required to be filed. The registrant agrees to furnish supplementally a copy of any omitted schedule or exhibit to the SEC upon request.
| Exhibit 1.67 | NOVALIQ Inventory (Omitted) | |
| Exhibit 1.68 | NOVALIQ Know-How (Omitted) | |
| Exhibit 1.70 | NOVALIQ Patent Rights (Omitted) | |
| Exhibit 3.5(b) | NOVALIQ Support Services (Omitted) | |
| Exhibit 4.3 | Validation Batches Responsibilities (Omitted) | |
| Exhibit 5.3 | Marketing Plan (Omitted) |
| 2 |
License Agreement
This License Agreement (the “Agreement”) is entered into on June 6, 2023 (“Signing Date”)
BY AND BETWEEN
NOVALIQ GMBH, a company organised under the laws of Germany and having its representative office at Im Neuenheimer Feld 515, 69120 Heidelberg, Germany (“NOVALIQ”)
AND
HARROW HEALTH, Inc. a company organised under the laws of Delaware and having its representative office at 102 Woodmont Blvd., Suite 610, Nashville, TN 37205, United States (“Harrow Health”), Harrow Eye, LLC, a limited liability company formed under the laws of Delaware and located at 102 Woodmont Blvd. Suite 610, Nashville, Tennessee 37205 United States (“Harrow Eye”) and Harrow IP, LLC, a limited liability company formed under the laws of Delaware and located at 102 Woodmont Blvd. Suite 610, Nashville, Tennessee 37205 United States (“Harrow IP”) (Harrow Health, Harrow Eye and Harrow IP being collectively referred to as “HARROW”).
RECITALS
WHEREAS, NOVALIQ is an ophthalmic pharmaceutical company with the mission to transform ocular therapeutics based on its topical anti-inflammatory and immunomodulating ophthalmic drug solution VEVYE®.
WHEREAS, HARROW is a pharmaceutical company focusing on the research, development, manufacturing and commercialization of pharmaceutical products in the ophthalmic market.
WHEREAS, HARROW owns IMPRIMIS Rx (“IMPRIMIS”), a division of HARROW whose primary business is directed to the compounding of ophthalmic products that are not formally approved by the FDA (as defined below) for the treatment or prevention of any specific disease.
WHEREAS, HARROW desires to obtain from NOVALIQ, and NOVALIQ is willing to grant to HARROW, an exclusive license under the NOVALIQ Technology for the development, manufacture and commercialization of the Licensed Product in the Field in the Territory (all such capitalized terms as defined below) on the terms and conditions of this Agreement.
NOW, THEREFORE, the Parties hereby agree as follows:
| 3 |
| 1. | DEFINITIONS |
For purposes of this Agreement, the following capitalized terms shall have the following meanings, whether used in the singular or plural:
| 1.1 | “Affiliate” means, with respect to a Party, any corporation or other legal entity other than that Party in whatever country organized, controlling, controlled by or under common control with that Party. For the purpose of this Section 1.1, the term “control” shall mean the ownership of more than fifty per cent (50%) of the equity share capital of an entity having the right to vote to elect directors of any such entity, or equivalent control in the case of an entity without share capital; provided, however, that regarding NOVALIQ, Affiliate shall not include Mr. Dietmar Hopp, direct relatives of Mr. Dietmar Hopp, dievini Hopp BioTech holding GmbH & Co. KG and/or any other companies controlled by Mr. Dietmar Hopp, his direct relatives and/or dievini Hopp BioTech holding GmbH & Co.KG that are not subsidiaries of NOVALIQ. |
| 1.2 | “Alliance Manger” shall have the meaning as set forth in Section 8.4(a). |
| 1.3 | “Applicable Laws” means any and all applicable federal, state, local, and international laws, rules and regulations, including without limitation Regulatory Authority regulations and environmental laws, as amended from time to time, and the regulations promulgated thereunder, as amended from time to time. |
| 1.4 | “Auditor” shall have the meaning as set forth in Section 7.5(b). |
| 1.5 | “Breaching Party” shall have the meaning as set forth in Section 14.3. |
| 1.6 | “Business Day(s)” means a day (other than Saturday or Sunday) on which banks are open for business in Heidelberg, Germany, and in Nashville, TN, United States. |
| 1.7 | “Calendar Quarter(ly)” means the respective periods of three (3) consecutive calendar months ending on March 31, June 30, September 30 or December 31, for so long as this Agreement is in effect, provided that (i) the first Calendar Quarter during the term of this Agreement shall extend from the Effective Date to the end of the first full Calendar Quarter thereafter; and (ii) the last Calendar Quarter during the term of this Agreement shall end upon the termination or expiry of this Agreement. |
| 1.8 | “Calendar Year(s)” means the respective periods beginning on January 1 and ending on December 31 of the same year, for so long as this Agreement is in effect, provided that (i) the first Calendar Year during the term of this Agreement shall extend from the Effective Date and end on December 31, 2023; and (ii) the last Calendar Year during the term of this Agreement shall end upon the termination or expiry of this Agreement. |
| 1.9 | “Change of Control” means, with respect to an entity (a) a transaction or series of related transactions that result(s) in the sale, license, transfer or other disposition of all or substantially all of such entity’s assets to a Third Party; or (b) a merger with a Third Party or other comparable consolidation with a Third Party in which such entity is not the surviving corporation or in which, if such entity is the surviving corporation, the shareholders of such entity immediately prior to the consummation of such merger or consolidation do not, immediately after consummation of such merger or consolidation, possess, directly or indirectly through one or more intermediaries, a majority of the voting power of all of the surviving entity’s outstanding stock and other securities and the power to elect a majority of the members of such entity’s board of directors; or (c) a transaction or series of related transactions (which may include a tender offer for such entity’s stock or the issuance, sale or exchange of stock of such entity) if the shareholders of such entity immediately prior to the initial transaction do not, immediately after consummation of such transaction or any of such related transactions, own, directly or indirectly through one or more intermediaries, stock or other securities of the entity that possess a majority of the voting power of all of such entity’s outstanding stock and other securities and the power to elect a majority of the members of such entity’s board of directors. |
| 4 |
| 1.10 | “Change of Control Payment” shall have the meaning as set forth in Section 7.2(c). |
| 1.11 | “Claim” shall have the meaning as set forth in Section 13.3(a). |
| 1.12 | “Clinical Trial” means a clinical study of the Licensed Product involving the administration of Licensed Product to human subjects. |
| 1.13 | “Commercially Reasonable Efforts” means, (a) with respect to the efforts to be expended by any Party with respect to any objective, such reasonable, sustained, diligent, and good faith efforts as such Party would normally use to accomplish a similar objective under similar circumstances, and (b) with respect to any objective relating to Development, Manufacture or Commercialization of the Licensed Product by HARROW, its Affiliates or Sublicensees, the carrying out of such activities in a sustained and diligent manner consistent with the efforts as HARROW, its Affiliate or Sublicensees would normally devote to a compound or product of similar value, stage of development, and life cycle and market potential at a similar stage, but in any event at least using efforts and resources comparable to the efforts and resources commonly used by pharmaceutical companies of comparable size for internally developed compounds and products of similar market potential at a similar stage in development or product life. |
| 1.14 | “Commission” shall mean the Securities and Exchange Commission. |
| 1.15 | “CMO” means a Third Party contract Manufacturing organization performing Manufacturing services. |
| 1.16 | “Competing Product” shall have the meaning as set forth in Section 2.4(b). |
| 1.17 | “CRO” means a Third Party contract research organization performing Development services. |
| 1.18 | “Confidential Information” of a Party means any technical, business or other information provided by or on behalf of one Party (including its Affiliates) to the other Party (including its Affiliates) in connection with this Agreement, whether prior to, on or after the Signing Date, including information relating to the terms of this Agreement, information relating to any Product (including the regulatory documentation), any Development or Commercialization of any Product, or the scientific, regulatory or business affairs or other activities of either Party, including any Know-How that such Party discloses to the other Party under this Agreement, or otherwise becomes known to the other Party by virtue of this Agreement. For the avoidance of doubt, Confidential Information includes information provided by or on behalf of one Party to the other Party prior to the Signing Date and falling into the definition of “Confidential Information” under the Confidentiality Agreement. The terms and conditions of this Agreement shall be deemed Confidential Information of both Parties (and both Parties shall be deemed the receiving Party with respect thereto). |
| 1.19 | “Confidentiality Agreement” means the confidentiality agreement entered into by and between HARROW and NOVALIQ and effective as of December 6, 2022. |
| 1.20 | “Control” or “Controlled” means, with respect to Confidential Information, Patent Rights, Know-How, Trademarks, Development Data and Regulatory Data that a Party owns or has a license to use, practice grant access or sublicense such Confidential Information, Patent Rights, Know-How, Trademarks, Development Data or Regulatory Data as provided herein without violating the terms of any agreement or other arrangement with any Third Party and without violating Applicable Laws. |
| 5 |
| 1.21 | “DED” means dry eye disease (e.g., keratoconjunctivitis sicca; ICD-10-CM Diagnosis Code H16.22), including any other disease or condition that is associated with dry eye disease for which the Product is indicated for treating or preventing under the approved FDA or Health Canada label. For clarity, DED does, inter alia, not include the following diseases: |
| (a) | blepharitis: anterior, or posterior, inflammatory or infectious (bacteria/ virae/ fungi/ mites), obstructive hair follicles or obstructive meibomian glands, often referred to as “eyelid syndrome”; |
| (b) | Hordeolum and chalazion; |
| (c) | Keratoconjunctivitis, conjunctivitis, keratitis etc. with bacteria, virae, fungi, parasites, utilizing (i) anti-bacterial, fungicide, anti-viral or anti-parasitic agents or utilizing (ii) antiseptic agents |
| (d) | Vernal keratoconjunctivitis; and |
| (e) | ocular hypersensitivity (itching) associated with ocular allergy (i.e., allergic conjunctivitis). |
| 1.22 | “Development” means all non-clinical and clinical research and drug development activities conducted with respect to the Licensed Product, including those necessary to obtain or maintain Regulatory Approvals (including reimbursement approval) and to successfully Manufacture and Market the Licensed Product for use in the Field. “Development” shall include, without limitation, chemistry, manufacturing and control (CMC), test method development and stability testing, formulation development, delivery system development, non-clinical testing including (clinical) virology, mechanism studies, toxicology, pharmacokinetics, Clinical Trials (whether conducted prior to, or post Regulatory Approvals), quality assurance/quality control, regulatory affairs activities, statistical analysis and report writing, submission of documents, market research and pharmacoeconomic studies. “Develop” shall have a correlative meaning. |
| 1.23 | “Development Data” means any pre-clinical and clinical data and any other data relating to the Development of the Licensed Product in the Field, and any report, document or other documentation containing or embodying such data. |
| 1.24 | “Disclosing Party” shall have the meaning as set forth in Section 11.1. |
| 1.25 | “Effective Date” shall mean the earlier to occur (i) December 2, 2023 or (ii) the receipt by HARROW of a written notice from NOVALIQ in which NOVALIQ confirms that NOVALIQ has the full right, power and authority to grant the license under Section 2.1. |
| 1.26 | “Executive Officers” means for NOVALIQ, the Chief Executive Officer of NOVALIQ and for HARROW, the Chief Executive Officer of HARROW. |
| 1.27 | “F4H5” means 1-perfluorobutylpentane. |
| 1.28 | “FDA” means the United States Food and Drug Administration, or a successor agency thereto. |
| 1.29 | “Field” means all uses of Licensed Product for the treatment of DED, in humans. |
| 1.30 | “First Commercial Sale” means the first sale of the Licensed Product to a Third Party on arm’s length terms by HARROW, its Affiliate or Sublicensee for use in the Field in the Territory, after the Licensed Product has been granted Marketing Authorization, provided, however, that in the event of a sale of the Licensed Product prior to Marketing Authorization which is substantially comparable to a commercial sale effected only after Marketing Authorization is obtained, then the first sale in any such arrangement shall also constitute a First Commercial Sale. |
| 1.31 | “First Upfront Payment” shall have the meaning as set forth in Section 7.1(a). |
| 6 |
| 1.32 | “FTE” means a full time equivalent person-year based upon a total of one thousand six hundred sixty (1,660) working hours per Calendar Year of work carried out by a duly qualified employee of NOVALIQ on or directly related to the work to be conducted under the Agreement. The portion of a FTE billable by NOVALIQ for one (1) individual during a given accounting period shall be determined by dividing the number of hours worked by said individual on the work to be conducted under the Agreement during such accounting period and the number of FTE hours applicable for such accounting period based on one thousand six hundred sixty (1,660) working hours per Calendar Year. |
| 1.33 | “FTE Rate” shall mean [***] per year. The FTE Rate includes all internal overhead and costs of internal consumables, unless agreed otherwise between the Parties. |
| 1.34 | “HARROW Cancer Drug Candidate” shall have the meaning as set forth in Section 9. |
| 1.35 | “HARROW Claim” shall have the meaning as set forth in Section 13.1. |
| 1.36 | “HARROW Indemnitee” shall have the meaning as set forth in Section 13.1. |
| 1.37 | “HARROW Technology” means (i) any and all Patent Rights and Know-How Controlled by HARROW, its Affiliates or Sublicenses on the Effective Date or during the term of this Agreement and required or used for the Development, Manufacture and/or Marketing of the Licensed Product; and (ii) trademarks and designs Controlled by HARROW, its Affiliates or Sublicensees on the Effective Date or during the term of this Agreement and required or used for the Marketing of the Licensed Product. |
| 1.38 | “ICC” shall have the meaning set forth in Section 16.5(a). |
| 1.39 | “IMPRIMIS” shall have the meaning set forth in the preamble. |
| 1.40 | “ImprimisRx Pharmacy” means the ophthalmic pharmacy operated by HARROW. |
| 1.41 | “Improvements” means: (a) any and all ideas, information, Know-How, data, research results, writings, inventions, discoveries, modifications, enhancements, derivatives, new uses, developments, techniques, materials, compounds, products, designs, processes, or other technology or intellectual property rights, whether or not patentable or copyrightable, in each case, that involves the use of the Licensed Product and that is an improvement to then-existing Novaliq Technology for the Licensed Product, and is developed by HARROW, its Affiliates, or Third Parties acting on its behalf while performing activities under this Agreement; and (b) all Patent Rights and other intellectual property rights in any of the foregoing. |
| 1.42 | “Improvement Patent Right” means a Patent Right covering an Improvement. |
| 1.43 | “Invention” means any process, method, composition of matter, article of manufacture, discovery, use or finding that is conceived and/or reduced to practice in the performance of activities under this Agreement. |
| 1.44 | “Joint Inventions” shall have the meaning as set forth in Section 10.1(a). |
| 1.45 | “Joint Know-How” shall have the meaning as set forth in Section 10.1(a). |
| 1.46 | “Joint Patent Rights” shall have the meaning as set forth in Section 10.1(a). |
| 1.47 | “Joint Steering Committee” and “JSC” shall have the meaning as set forth in Section 8.1(a). |
| 1.48 | “Klarity C” means the product made by IMPRIMIS that contains 0.1% cyclosporine in an ophthalmic emulsion for topical use. |
| 7 |
| 1.49 | “Klarity C plus Loteprednol” means the product made by IMPRIMIS that contains 0.1% cyclosporine and 0.2% loteprednol etabonate in an ophthalmic emulsion for topical use. |
| 1.50 | “Klarity Drops” means the product made by IMPRIMIS that has the same aqueous emulsion delivery vehicle as Klarity C but, except for glycerin, does not contain any other active ingredient, including cyclosporine. |
| 1.51 | “Know-How” means all technical, scientific and other information, inventions, discoveries, trade secrets, knowledge, technology, means, methods, processes, practices, formulae, instructions, skills, techniques, procedures, expressed ideas, technical assistance, designs, drawings, assembly procedures, computer programs, apparatuses, specifications, Development Data, Regulatory Data, results, non-clinical, clinical, safety, process and Manufacturing and quality control data and information (including trial designs and protocols), registration dossiers, in each case, solely to the extent confidential and proprietary and in written, electronic or any other form now known or hereafter developed. |
| 1.52 | “Knowledge” shall have the meaning as defined in Section 12.2. |
| 1.53 | “Licensed Product” means a finished product which is a composition consisting or consisting essentially of cyclosporine A (CAS-Number: 59865-13-3) dissolved in a vehicle of 1-perfluorobutylpentane (F4H5) and up to 1 wt.% ethanol. For purposes of this Section 1.53, “consisting of” shall mean that no further features (components) are present apart from the ones following said wording. In the case of compositions, the use of “consisting essentially of” means that specific further components can be present, namely those not materially affecting the essential characteristics of the composition. For the avoidance of doubt, Licensed Product shall include the finished dosage form product formulation described in NDA 217469. |
| 1.54 | “Losses” shall have the meaning as set forth in Section 13.1. |
| 1.55 | “Manufacturing” means, with respect to Licensed Product, the handling and storage of substances, and other materials, the manufacturing, processing, filling, finishing, packaging and labeling (excluding the development of packaging and labeling components for Regulatory Approval), holding (including storage), stability testing, quality assurance and quality control testing (including release) of the Licensed Product (other than quality assurance and quality control related to the Development of the Manufacturing process, which activities shall be considered Development activities) and shipping of the Licensed Product. “Manufacture” shall have a correlative meaning. |
| 1.56 | “Marketing” means any and all activities directed to the offering for sale and sale of the Licensed Product, both before (to the extent permitted under Applicable Laws) and after Marketing Authorization has been obtained, including activities related to promoting, marketing, distributing, selling, providing support for the Licensed Product, placing and making the Licensed Product available on the Market. “Market” shall have a correlative meaning. |
| 1.57 | “Marketing Authorization” means, with respect to a given pharmaceutical product and a given country or jurisdiction, receipt of approval of an NDA for the commercial marketing of such product from the applicable Regulatory Authority(ies) in such country or jurisdiction, but excluding any Pricing or Reimbursement Approval. |
| 1.58 | “Marketing Plan” shall have the meaning as set forth in Section 5.3. |
| 1.59 | “Milestone Event” shall have the meaning as set forth in Sections 7.2(a) and 7.2(b). |
| 1.60 | “Milestone Notice” shall have the meaning as set forth in Section 7.2(a). |
| 8 |
| 1.61 | “NDA” means a New Drug Application filed pursuant to the requirements of the FDA, as more fully defined in 21 U.S. CFR § 314.3 et seq., and any equivalent application submitted in any country in the Territory, including all additions, deletions or supplements thereto, and as any and all such requirements may be amended, or supplanted, at any time. |
| 1.62 | “Net Sales” means the gross invoice price of Licensed Product sold by HARROW or its Affiliates or Sublicensees to the first Third Party, less the following deductions if and to the extent such deductions to unaffiliated entities are actually allowed and granted: |
| (a) | trade, quantity, and/or cash discounts, charge-back payments, allowances or rebates, including promotional or similar discounts or rebates, and discounts or rebates to drug wholesaler, pharmacies, governmental or managed care organizations; |
| (b) | discounts provided in connection with coupon, voucher or similar patient programs; |
| (c) | credits or allowances given or made with respect to Licensed Product by reason of rejection, defects, recalls, returns, rebates, or retroactive price reductions; |
| (d) | bad debt actually written off during the accounting period (provided, however, that such deduction may not exceed one percent (1%) of the gross sales in the corresponding accounting period and that any bad debt write-off so deducted which is later reversed shall be added back to Net Sales in the accounting period in which the reversal occurs); |
| (e) | any tax, tariff, duty or government charge (including any sales, value added, excise or similar tax or government charge, but excluding any income tax) levied on the sale, transportation or delivery of Licensed Product and borne by HARROW, its Affiliates or Sublicensees without reimbursement from any Third Party; |
| (f) | any administrative fees paid to Third party group purchasing organizations or managed care entities for the sale of Licensed Product (provided, however, that such deduction may not exceed two percent (2%) of the gross sales in the corresponding accounting period); and |
| (g) | the value (i.e., [***]) of any unsold NDA Validation Batches of the Licensed Product. |
All such discounts, allowances, credits, rebates and other deductions shall be fairly and equitably allocated to the sale of the relevant Licensed Product by HARROW, its Affiliates or Sublicensees, such that the Licensed Product does not bear a disproportionate portion of such deductions as compared to other products sold separately from but with a certain link or other connection to the Licensed Product. For the avoidance of doubt, the Net Sales shall be calculated only once for the first bona fide arm’s length sale of the Licensed Product by either HARROW, its Affiliate or its Sublicensee, to a Third Party which is neither an Affiliate nor a Sublicensee of HARROW. Net Sales shall be determined in accordance with the accrual method of accounting in accordance with U.S. GAAP applied in a consistent manner. For the conversion of Net Sales from other currencies to US Dollars, the standard exchange rate methodology as applied in HARROW’s external reporting and accepted by HARROW’s auditor for such reporting shall be used.
| 1.63 | “Non-Breaching Party” shall have the meaning as set forth in Section 14.3. |
| 1.64 | “NOV03 Product” means any pharmaceutical, medical device, or over the counter product comprising: (a) a composition consisting or essentially consisting of 1-perfluorohexyloctane (F6H8; CAS-Number: 1 3333 1 -77-8); or (b) a composition consisting or consisting essentially of 1-perfluorohexyloctane, and one or more excipients; or (c) a composition consisting or consisting essentially of an omega-3 fatty acid ethylester, selected from docosahexaenoic acid ethyl ester, eicosapentaenoic acid ethyl ester or mixtures thereof, dissolved in a vehicle consisting or consisting essentially of 1-perfluorohexyloctane, and optionally one or more excipients. For purposes of this Section 1.64, “consisting of” shall mean that no further components are used or are present apart from the ones following said wording. In the case of compositions, “consisting essentially of” shall mean that further components can be included, provided that those components do not materially affect the essential characteristics of the composition. |
| 9 |
| 1.65 | “NOVALIQ Claim” shall have the meaning as set forth in Section 13.2. |
| 1.66 | “NOVALIQ Indemnitee” shall have the meaning as set forth in Section 13.2. |
| 1.67 | “NOVALIQ Inventory” means the inventory listed on Exhibit 1.67. |
| 1.68 | “NOVALIQ Know-How” means any and all Know-How Controlled by NOVALIQ or its Affiliates as of the Effective Date and during the Term of this Agreement that is necessary or reasonably useful for HARROW to Develop, Manufacture or Market the Licensed Product in the Field in the Territory. For the avoidance of doubt, NOVALIQ Know-How shall include NOVALIQ’s share in any Joint Know-How. Without limiting the generality of the definition set forth in this Section 1.68, the NOVALIQ Know-How in existence on the Signing Date is listed in Exhibit 1.68. For clarity, the NOVALIQ Know-How includes the NOVALIQ Manufacturing Know-How. |
| 1.69 | “NOVALIQ Manufacturing Know-How” means that part of the NOVALIQ Know-How that is required or reasonably useful for HARROW to Manufacture the Licensed Product in the Field. |
| 1.70 | “NOVALIQ Patent Rights” means the NOVALIQ Platform Patent Rights and the NOVALIQ Product Patent Rights. |
| 1.71 | “NOVALIQ Platform Patent Rights” means the Patent Rights (including NOVALIQ’s share in Joint Patent Rights) Controlled by NOVALIQ or any of its Affiliates on the Effective Date or during the term of this Agreement that claim or cover the Licensed Product in the Field in the Territory or which are required to Develop, Manufacture or Market the Licensed Product in the Field in the Territory, excluding the NOVALIQ Product Patent Rights; provided however, that NOVALIQ Platform Patent Rights do not include Patent Rights that may be Controlled by NOVALIQ in the future as a result of a Change of Control in NOVALIQ but which were Developed by a Third Party that caused NOVALIQ to undergo the Change of Control, prior to such Change of Control, or by NOVALIQ after such Change of Control wherein such Patent Rights claim priority to Patent Rights that were owned or Controlled by such Third Party prior to such Change of Control. Without limiting the generality of the definition set forth in this Section 1.71, the NOVALIQ Patent Rights in existence on the Signing Date are listed in Exhibit 1.70. |
| 1.72 | “NOVALIQ Product Patent Rights” means the Patent Rights (including NOVALIQ’s share in Joint Patent Rights) Controlled by NOVALIQ or any of its Affiliates on the Effective Date or during the term of this Agreement that claim or otherwise cover solely the Licensed Product in the Field in the Territory and no other product; provided however, that NOVALIQ Product Patent Rights do not include Patent Rights that may be Controlled by NOVALIQ in the future as a result of a Change of Control in NOVALIQ but which were Developed by a Third Party that caused NOVALIQ to undergo the Change of Control, prior to such Change of Control, or by NOVALIQ after such Change of Control wherein such Patent Rights claim priority to Patent Rights that were owned or Controlled by such Third Party prior to such Change of Control. Without limiting the generality of the definition set forth in this Section 1.71, the NOVALIQ Product Rights in existence on the Signing Date are listed in Exhibit 1.70. |
| 10 |
| 1.73 | “NOVALIQ Technology” means the NOVALIQ Patent Rights and the NOVALIQ Know-How. |
| 1.74 | “NOVALIQ Trademark” means (a) the VEVYE (word) trademark registration, US Reg. No. 6797943, the VEVYE (word/device) trademark application, US Serial No. 97622981, and any other trademark registration or trademark application1 containing the element VEVYE that is Controlled by NOVALIQ in the Territory during the Term (the “VEVYE Trademarks”); and (b) the EYESOL (word) trademark registration, US Reg. No. 5742345, the EYESOL (word/device) trademark registration, US Reg. No. 5790514, and any other trademark registration, trademark application containing the element EYESOL that is Controlled by NOVALIQ in the Territory during the Term. |
| 1.75 | “Party” means either NOVALIQ or HARROW and “Parties” means NOVALIQ and HARROW. |
| 1.76 | “Patent Right” means any patents and patent applications, together with all additions, divisions, continuations, continuations-in-part, substitutions, reissues, re-examinations, extensions, confirmations, registrations, supplemental protection certificates and renewals of any of the foregoing, as applicable in a given country or territory. |
| 1.77 | “PDUFA Fees” means all fees that are payable under the United States Prescription Drug User Fee Act or any comparable law in Canada. |
| 1.78 | “Pharmacovigilance Agreement” shall have the meaning as set forth in Section 3.8. |
| 1.79 | “Phase IV Clinical Trial” means a human clinical trial which is conducted on a product after Regulatory Approval of the product has been obtained from an appropriate Regulatory Authority, and includes (a) trials conducted voluntarily for enhancing marketing or scientific knowledge of an approved Indication or (b) trials conducted after Regulatory Approval due to request or requirement of a Regulatory Authority or as a condition of a previously granted Regulatory Approval. |
| 1.80 | “PRC” means the People’s Republic of China (including the Special Administration Zone Hong Kong and the Special Administration Zone Macao) and Taiwan. |
| 1.81 | “Pricing or Reimbursement Approval” means, with respect to a given pharmaceutical product and a given country or jurisdiction, an approval, agreement, determination, or other decision by the applicable Regulatory Authority that establishes prices charged to end-users that such product will be reimbursed in such country or jurisdiction, or any other approvals related to pricing, reimbursement or access to a pharmaceutical product (including all activities related to tenders and contracts). |
| 1.82 | “Qualified F4H5 Supplier” means [***]. |
| 1.83 | “Receiving Party” shall have the meaning as set forth in Section 11.1. |
| 1.84 | “Regulatory Approval” means, with respect to a given pharmaceutical product and a given country or jurisdiction, any and all approvals, licenses, registrations, determinations or authorizations of the relevant Regulatory Authority necessary for the development, manufacture or marketing of such product in such country or jurisdiction, including any Marketing Authorization or Pricing, post-approval variations or Reimbursement Approval, as applicable. |
| 1.85 | “Regulatory Authority(ies)” means any national, supra-national, regional, state or local regulatory agency, department, bureau, commission, council or other governmental entity in the Territory involved in the granting of Regulatory Approvals for the Development, Manufacturing, Marketing of the Licensed Product, including the FDA and Health Canada, or any successors. |
| 11 |
| 1.86 | “Regulatory Data” means all regulatory, technical, scientific or other data required or reasonably useful for obtaining or maintaining Regulatory Approvals for the Licensed Product in the Field, and any report, documents or other documentation containing or embodying such data, including (a) any NDA or other regulatory filing and regulatory dossiers; (b) any communications with Regulatory Authorities (including minutes and official contact reports) and all supporting documents with respect thereto, including all adverse event files and complaint files; and (c) clinical and other data contained or relied upon in any of the foregoing; in each of (a), (b) and (c) relating to the Licensed Product in the Field. |
| 1.87 | “ROFN” shall have the meaning as set forth in Section 9. |
| 1.88 | “ROFN Exercise Period” shall have the meaning as set forth in Section 9. |
| 1.89 | “ROFN Negotiation Period” shall have the meaning as set forth in Section 9. |
| 1.90 | “ROFN Transaction” shall have the meaning as set forth in Section 9. |
| 1.91 | “Royalty Term” means on a country-by-country basis, the period of time beginning with the First Commercial Sale of the Licensed Product in a country of the Territory and ending with the later of: (a) the expiration of the last Valid Patent Claim of the NOVALIQ Patent Rights; or (b) the expiration of regulatory exclusivity for the Licensed Product in the Territory; or (c) twelve (12) years from the First Commercial Sale of the Licensed Product in such country of the Territory. |
| 1.92 | “ROW” means all countries worldwide other than the Territory. |
| 1.93 | “Second Upfront Payment” shall have the meaning as set forth in Section 7.1(b). |
| 1.94 | “Siegfried Irvine MSA” means the Master Services Agreement between NOVALIQ and Alliance Medical Products, Inc (d/b/a Siegfried Irvine) dated August 1, 2017, as subsequently amended by amendment no. 1, amendment no. 2 and amendment no. 3. |
| 1.95 | “Siegfried Irvine “ Medical Products, Inc (d/b/a Siegfried Irvine). |
| 1.96 | “Signing Date” shall have the meaning set forth in the preamble. |
| 1.97 | “Sublicensee” means any Third Party (other than Third Party contractors used by HARROW in the Development, Manufacture and Marketing of the Licensed Product on HARROW’s behalf in accordance with Section 3.9) obtaining a sublicense under the NOVALIQ Technology in accordance with Section 2.2. |
| 1.98 | “Tax” means any kind of direct or indirect tax, including but not limited to any income, profits, capital gains, sales, use, ad valorem, receipts, value added, goods and services, license, withholding, excise, property, net worth, transfer or stamp taxes, customs, duties and charges and any other assessment or governmental charge, together with all interest, penalties and additions imposed with respect to such amounts. |
| 1.99 | “Term” shall have the meaning as set forth in Section 14.1. |
| 1.100 | “Territory” means the United States of America and Canada (including their respective territories and possessions). |
| 12 |
| 1.101 | “Third Party” means any person or entity other than NOVALIQ, HARROW and their respective Affiliates. |
| 1.102 | “Third Party Allegation” shall have the meaning as set forth in Section 10.4(a). |
| 1.103 | “Third Party Infringement” shall have the meaning as set forth in Section 10.3(a). |
| 1.104 | “U.S. MA” shall have the meaning as set forth in Section 3.2(a). |
| 1.105 | “Validation Batch” means the validation batches of Licensed Product manufactured by Siegfried Irvine under the Siegfried Irvine MSA. |
| 1.106 | “Valid Patent Claim” means a claim (a) of any patent application within the NOVALIQ Patent Rights pending in the Territory that has not been pending for more than ten (10) years from the earliest filing date to which such claim or the applicable patent application is entitled to claim priority and which claim has not been revoked, cancelled, withdrawn, held invalid or abandoned, provided further that, if said pending claim with a pendency of at least ten (10) years or longer subsequently issues, it will be considered a Valid Claim upon issuance; or (b) of any issued, unexpired Patent Right within the NOVALIQ Patent Rights in the Territory that has not been held finally invalid or unenforceable by a court or administrative body of competent jurisdiction in an unappealed or unappealable decision. |
| 1.107 | “VEVYE Trademarks” shall have the meaning as set forth in Section 1.74. |
| 1.108 | Interpretation. In this Agreement, unless otherwise specified: |
| (a) | the word “including” or any variation thereof shall mean “including without limitation” or any variation thereof and shall not be construed to limit any general statement which it follows to the specific or similar items or matters immediately following it; |
| (b) | the word “or” will not be exclusive, unless the context otherwise requires; |
| (c) | words denoting the singular shall include the plural and vice versa and words denoting any gender shall include all genders; |
| (d) | words such as “herein”, “hereof”, and “hereunder” refer to this Agreement as a whole and not merely to the particular provision in which such words appear; and |
| (e) | the Exhibits and other attachments form part of the operative provision of this Agreement and references to this Agreement shall include references to the Exhibits and attachments. |
| 2. | Grant and Scope of License; Exclusivity |
| 2.1 | License Grant |
Subject to the terms and conditions of this Agreement, effective as of the Effective Date, NOVALIQ hereby grants to HARROW, and HARROW hereby accepts from NOVALIQ, a license, with the right to grant sublicenses (subject to the restrictions set forth in Section 2.2), under the NOVALIQ Technology and the NOVALIQ Trademarks to: (i) further Develop and Market the Licensed Product in the Field in the Territory, which license shall be exclusive (even as to NOVALIQ and its Affiliates), and (ii) Manufacture the Licensed Product in the Territory and in the ROW, excluding the PRC, (for the sole purpose of Developing and Marketing the Licensed Product in the Field in the Territory), which license shall be non-exclusive.
| 13 |
| 2.2 | Sublicensing |
| (a) | HARROW shall have the right to sublicense its rights under Section 2.1: (i) to any of its Affiliates without NOVALIQ’s consent, provided that it shall inform NOVALIQ of such sublicense, and (ii) to Third Parties (except sublicenses to CROs or to CMOs for the Development or Manufacture of Licensed Product) with NOVALIQ’s prior written consent. |
| (b) | The right to sublicense is subject to a written sublicense agreement containing terms and conditions that are consistent with those contained in this Agreement, and shall include, inter alia, provisions regarding confidentiality, non-compete, indemnification, audit, record-keeping, termination and consequences of termination for NOVALIQ’s protection that are consistent with the corresponding terms and conditions provided herein. HARROW shall remain liable to NOVALIQ for all obligations under this Agreement, including all payment obligations, and shall send to NOVALIQ a copy of the executed sublicensing agreement within twenty (20) Business Days after its execution. |
| 2.3 | Retained Rights; No Implied Licenses |
NOVALIQ retains all rights under the NOVALIQ Technology (a) outside the Field in the Territory and (b) (inside and outside the Field) in the ROW, and NOVALIQ shall be permitted to Develop and Manufacture the Licensed Product within the Territory, solely to Market the Licensed Product outside the Field in the Territory or (inside and outside the Field) in the ROW. No right or license is granted to either Party hereunder by implication, estoppel, or otherwise to any Know-How, Patents or other intellectual property right owned or otherwise Controlled by the other Party or its Affiliates, except as expressly set forth in this Agreement.
| 2.4 | Non-competition |
| (a) | With the exception of any NOV03 Product, during the Term, NOVALIQ and its Affiliates shall not develop or market any product for the treatment of DED in the Territory. If NOVALIQ undergoes a Change of Control, the provisions of this Section 2.4(a) shall not apply to any product which (i) is developed by a party that is an Affiliate of NOVALIQ as a result of such Change of Control but which was not an Affiliate of NOVALIQ prior to such Change of Control, and (ii) is not developed utilizing technology Controlled by NOVALIQ or Controlled by a party which would have been considered a NOVALIQ Affiliate prior to such Change of Control. |
| (b) | With the exception of Klarity Drops, Klarity C and Klarity C plus Loteprednol, during the Term, HARROW, its Affiliates or Sublicensees holding rights to the Licensed Product in the Field in the Territory shall not develop or market any prescription product for the treatment of DED in the Territory. HARROW further agrees that it will not seek to expand the product label indications in the Territory for any prescription product it markets and falls within one or more of the exceptions (a) – (e) as set forth in Section 1.20 to include the treatment of DED. Notwithstanding the above, it shall not be a violation of this Section 2.4(b) for HARROW to maintain its passive minority investment interest in Surface Ophthalmics Inc. |
| (c) | If a Party acquires (whether by merger, purchase of assets or equity, or otherwise), or if as a result of a Change of Control (where it is the controlling party), owns, a product that has obtained Regulatory Approval in the Field in the Territory, and whose commercialization would violate Section 2.4(a) or Section 2.4(b), as applicable (“Competing Product”), such Party shall, within thirty (30) calendar days after such acquisition or Change of Control, or, if such notification is not possible within such period due to confidentiality restrictions or Applicable Laws, as soon as practicable following such acquisition or Change of Control, notify the other Party of such acquisition or Change of Control. Furthermore, such Party shall divest or terminate its rights to commercialize such Competing Product in the Field in the Territory, and shall, use Commercially Reasonable Efforts to divest or terminate its rights to commercialize such Competing Product within twelve (12) months after such acquisition or Change of Control has occurred provided that this Agreement is still in effect as of such date. Solely with respect to a Competing Product, as long as such Party complies with its obligations under this Section 2.4(c), such Party shall not be in breach of Section 2.4(a) or Section 2.4(b), as applicable, with respect to such Competing Product |
| 14 |
| 3. | Know-How Transfer; DEVELOPMENT; Regulatory |
| 3.1 | Transfer of NOVALIQ Know-How |
Within a period of ninety (90) days following the Effective Date, NOVALIQ shall (a) provide to HARROW, in a mutually-agreed upon format, the Clinical Data, Regulatory Data and other material Know-How included in the NOVALIQ Know-How, with the exception of the NOVALIQ Manufacturing Know-How which will be transferred in accordance with Section 4; and (b) make its relevant personnel reasonably available to HARROW, at reasonable times during NOVALIQ’s normal business hours and upon reasonable prior notice, to answer any questions or provide instructions as reasonably requested by HARROW concerning the information delivered pursuant to this Section 3.1. Thereafter, any support by NOVALIQ will be reimbursed by HARROW under Section 3.5.
| 3.2 | Marketing Authorization for the United States |
| (a) | The Parties acknowledge that NOVALIQ filed with the FDA an NDA for the Licensed Product for the United States and the FDA approved the Licensed Product on May 30, 2023 (the “U.S. MA”). |
| (b) | As soon as reasonably practicable following the Effective Date, NOVALIQ will assign and transfer to HARROW, and HARROW will assume from NOVALIQ, the U.S. MA, and all of NOVALIQ’s rights, interest and title therein. NOVALIQ and HARROW each agree to use Commercially Reasonable Efforts to take all actions required by the FDA to effect the transfer of the U.S. MA from NOVALIQ to HARROW. |
| (c) | Until such time as the U.S. MA is effectively transferred and assigned to HARROW, NOVALIQ shall be responsible for interfacing, corresponding and meeting with the FDA with respect to the Licensed Product, provided that NOVALIQ shall inform HARROW of any meetings and material correspondence with the FDA reasonably in advance and shall provide copies of such material correspondence with the FDA to HARROW, and HARROW shall have the right to attend any such meeting with the FDA as an observer. |
| (d) | Following the effective transfer of the U.S. MA, and except as set forth in the second sentence of Section 3.4(a), HARROW shall (a) be responsible for obtaining and maintaining all Regulatory Approvals for the Licensed Product for the United States, at its sole cost and expense; (b) shall be responsible for interfacing, corresponding and meeting with the FDA with respect to the Licensed Product, at its sole cost and expense; provided that HARROW shall inform NOVALIQ of any meetings and material correspondence with the FDA reasonably in advance and shall provide copies of any such material correspondence with the FDA to NOVALIQ, and NOVALIQ shall have the right to attend any such meeting with the FDA as an observer. |
| 15 |
| 3.3 | Marketing Authorization for Canada |
| (a) | HARROW shall be solely responsible for obtaining and maintaining all Regulatory Approvals for the Licensed Product in Canada and for interfacing, corresponding and meeting with Health Canada with respect to the Licensed Product, at its sole cost and expense. HARROW shall use Commercially Reasonable Efforts to file an NDA with Canada Health within two (2) years following the Effective Date. |
| (b) | HARROW shall inform NOVALIQ of any meetings and material correspondence with Health Canada reasonably in advance and shall provide copies of any such material correspondence with Health Canada to NOVALIQ, and NOVALIQ shall have the right to attend any such meeting with the FDA as an observer. |
| 3.4 | Further Development of Licensed Product |
| (a) | Subject to the terms and conditions of this Agreement, and except as set forth in the last sentence of this Section 3.4(a) HARROW shall have the sole right and responsibility to further Develop the Licensed Product in the Field in the Territory, at its sole cost and expense. Notwithstanding the above, NOVALIQ shall be solely responsible for conducting the post-marketing study that is identified as 4454-1 in the FDA’s NDA Approval Letter for the Licensed Product dated May 30, 2023, in accordance with the timeline submitted to the FDA on May 26, 2023. The Parties will comply with all regulatory requirements for submission, Clinical Trial conduction and reporting under IND 128 163. HARROW will supply NOVALIQ with the quantities of commercial Licensed Product required by NOVALIQ to conduct the post-marketing study at a price of [***] per unit. |
| (b) | HARROW shall conduct the further Development of the Licensed Product in the Field in the Territory in a way that the respective activities will likely not negatively impact the Development of the Licensed Product by NOVALIQ outside the Field in the Territory or (inside and outside the Field) in the ROW. |
| (c) | If HARROW further Develops the Licensed Product in the Field in the Territory, HARROW shall provide the JSC with a written report at the end of each Calendar Quarter summarizing in reasonable detail HARROW’s and its Affiliates’ material activities and progress related to the further Development of the Licensed Product in the Field in the Territory. |
| (d) | HARROW shall maintain records, in sufficient detail and in good scientific manner appropriate for patent and regulatory purposes, which shall fully and properly reflect all work done and results achieved under this Agreement. |
| 3.5 | Support by NOVALIQ |
| (a) | Upon HARROW’s request, NOVALIQ will use Commercially Reasonable Efforts to support HARROW in the further Development of the Licensed Product in the Field in the Territory, including with respect to regulatory matters. NOVALIQ and HARROW will agree on the scope of activities to be performed by NOVALIQ, the number of NOVALIQ FTEs to perform such activities, and the budget estimates to perform such activities before NOVALIQ initiates any work hereunder. |
| (b) | Without limiting Section 3.5(a), following the Effective Date, NOVALIQ will provide to HARROW the services listed in Exhibit 3.5(b) through the subcontractors listed in Exhibit 3.5(b). |
| 16 |
| (c) | HARROW shall compensate NOVALIQ for any support hereunder on an FTE basis at the FTE Rate and shall reimburse NOVALIQ for any out of pocket expenses at cost plus a mark-up of ten percent (10%) within thirty (30) days upon receipt of an invoice from NOVALIQ. |
| 3.6 | PDUFA Fees |
HARROW will pay any and all PDUFA Fees incurred for the Licensed Product for the Territory that become due on or after the Effective Date. For the avoidance of doubt, NOVALIQ shall be solely responsible for any fees that were due at the time of filing the NDA for the Licensed Product.
| 3.7 | Exchange and Use of Development Data and Regulatory Data |
| (a) | If during the Term, NOVALIQ obtains new Development Data or Regulatory Data that is required or reasonably useful for HARROW to obtain or maintain Regulatory Approvals for the Licensed Product in the Territory, NOVALIQ shall provide such Development Data or Regulatory Data, as applicable, to HARROW, and HARROW may use such Development Data or Regulatory Data, as applicable, as part of the NOVALIQ Know-How in accordance with the terms and conditions of this Agreement. |
| (b) | HARROW shall provide to NOVALIQ, free of charge, copies of all Development Data and Regulatory Data Controlled by HARROW during the Term promptly after such Development Data or Regulatory Data, as applicable, comes under the Control of HARROW. HARROW hereby grants to NOVALIQ the irrevocable, perpetual right and license (with the right to sublicense in multiple tiers) to use (including any referencing) the Development Data and Regulatory Data Controlled by HARROW for the Development, Manufacture and Marketing of the Licensed Product outside the Field in the Territory and (inside and outside the Field) in the ROW on a royalty-free basis, without the requirement of obtaining HARROW’s consent. HARROW shall provide such support to NOVALIQ and shall execute and deliver such instruments and do such acts and things as may be necessary under Applicable Law or as are requested by a Regulatory Authority or as NOVALIQ may reasonably request to enable NOVALIQ to make use of the Development Data or Regulatory Data, as applicable, under this Section 3.7(b). Furthermore, HARROW will allow Regulatory Authorities of the ROW to inspect the Development Data and Regulatory Data that are Controlled by HARROW. For support requested by NOVALIQ pursuant to this Section 3.7(b), NOVALIQ shall compensate HARROW on an FTE basis at the FTE Rate and shall reimburse HARROW for any out of pocket expenses at cost plus a mark-up of ten percent (10%) within thirty (30) days upon receipt of an invoice from HARROW. |
| 3.8 | Pharmacovigilance |
The Parties shall have in place and will maintain during the Term systems, procedures, training programs and documentation needed to perform and comply with their pharmacovigilance regulatory obligations, and each Party shall promptly inform the other Party of any safety issues that may arise and that need to be reported under applicable Laws. The Parties shall negotiate in good faith a pharmacovigilance agreement for the Licensed Product with customary terms and conditions to be negotiated in good faith and agreed upon by the Parties within ninety (90) days of the Effective Date (the “Pharmacovigilance Agreement”). Each Party will ensure that it complies with all applicable Laws regarding the Licensed Product relating to risk management, drug safety and pharmacovigilance. For the avoidance of doubt, any and all costs (including internal or external expenses of NOVALIQ) incurred under this Section 3.8 in relation to the Licensed Product in the Field in the Territory will be solely borne by HARROW (including a proportionate portion of the costs incurred for coordinating and liaising between all licensees of the Licensed Product inside and outside the Field and inside and outside of the Territory, e.g. for safety data exchange, etc.) and any and all costs incurred under this Section 3.8 solely in relation to the Licensed Product outside of the Field in the Territory or in the ROW will be solely borne by NOVALIQ.
| 17 |
| 3.9 | Subcontracting |
Each Party shall have the right to engage Third Party subcontractors to perform any of its obligations under this Agreement. Any such subcontractor shall meet the qualifications typically required by that Party for the performance of work similar in scope and complexity to the subcontracted activity and must be bound by confidentiality and non-use obligations no less stringent that those set out in this Agreement. A Party’s use of subcontractors shall not relieve it of any of its obligations pursuant to this Agreement. Any Party engaging a subcontractor to perform any of its obligations shall remain responsible for the performance of such obligations and shall be liable to the other Party for the acts and omissions of each of its subcontractors which would amount to a breach of this Agreement to the same extent as if such acts and omissions had been done by the subcontracting Party itself.
| 4. | MANUFACTURING |
| 4.1 | Manufacturing Responsibility |
Subject to Section 4.2 and Section 4.3, and effective as of the Effective Date HARROW shall have the sole right and responsibility, at its sole cost and expense, to Manufacture, by itself or through Affiliates or CMOs, Licensed Product in the Territory or in the ROW, excluding PRC, for the Marketing of such Licensed Product in the Field in the Territory. Upon the request of HARROW, NOVALIQ shall (a) use commercially reasonable efforts to liaise between HARROW and its qualified suppliers regarding a direct supply agreement between HARROW and such qualified suppliers; and (b) permit its qualified suppliers to Manufacture Licensed Product directly for HARROW for use in the Field in the Territory. In the event that neither (a) nor (b) in the preceding sentence is achieved, the Parties shall work together to determine the best way HARROW can obtain Licensed Product for further distribution in the Territory in the Field. Any costs and expenses incurred for a Manufacturing technology transfer from a qualified supplier currently used by NOVALIQ to a new supplier engaged by HARROW shall be borne by HARROW.
| 4.2 | 1-perfluorobutylpentane (F4H5) Supply |
| (a) | Subject to Section 4.2(b), HARROW shall obtain F4H5 exclusively from NOVALIQ’s Qualified F4H5 Suppliers. NOVALIQ will use commercially reasonable efforts to liaise between HARROW and the Qualified F4H5 Suppliers regarding a direct supply agreement between HARROW and such Qualified F4H5 Suppliers, and any such supply agreement shall be subject to NOVALIQ’s prior written consent, such consent not to be unreasonably withheld, conditioned or delayed. |
| (b) | HARROW shall have the right to Manufacture F4H5, by itself or through Affiliates or CMOs other than the Qualified F4H5 Suppliers, if the Qualified F4H5 Suppliers are unable or unwilling to supply HARROW with the quantities of F4H5 required by HARROW. |
| 4.3 | Supply of Validation Batches and Inventory |
| (a) | Effective as of the Effective Date, HARROW purchases from NOVALIQ, and NOVALIQ sells to HARROW, the NOVALIQ Inventory, which NOVALIQ Inventory shall include the Validation Batches. The Validation Batches shall be packaged in HARROW’s trade dress, which HARROW shall provide to NOVALIQ without unreasonable delay. NOVALIQ will deliver or will instruct and authorize Siegfried Irvine to deliver to HARROW the NOVALIQ Inventory in accordance with the estimated delivery dates set forth on Exhibit 1.67. Delivery will be made Ex Works Siegfried Irvine manufacturing site (Incoterms® 2020). In consideration for the sale and delivery of the NOVALIQ Inventory, HARROW will pay to NOVALIQ (a) an amount of [***] within ten (10) Business Days following the Effective Date for the NOVALIQ Inventory currently held at stock and delivered upon the Effective Date in accordance with Exhibit 1.67, and (b) the purchase price for the remaining portion of the NOVALIQ Inventory as set forth in Exhibit 1.67 within thirty (30) days upon delivery of such NOVALIQ Inventory and receipt of an invoice from NOVALIQ. |
| 18 |
| (b) | The Validation Batches and the NOVALIQ Inventory items are experimental in nature and are provided “as is”, without any warranties as to merchantability or fitness for a particular purpose. In case that any of the Validation Batches or any item of the NOVALIQ Inventory delivered to HARROW under this Section 4.3 is defective, NOVALIQ will assign to HARROW the rights that NOVALIQ has in respect of such defective Validation Batch or NOVALIQ Inventory item, as application, against the respective CMO or supplier (e.g. with respect to the Validation Batches, the rights that NOVALIQ may have against Siegfried Irvine under the Siegfried Irvine MSA), and this shall be the sole and exclusive remedy that HARROW has against NOVALIQ in respect of such defective Validation Batch or NOVAliq Inventory Item, as applicable. |
| (c) | Within thirty (30) days following the Effective Date, the Parties will enter into one or more quality agreement(s) with respect to the supply of Validation Batches and NOVALIQ Inventory. |
| 5. | Marketing |
| 5.1 | Marketing Responsibilities |
Effective as of the Effective Date and during the Term, HARROW will be solely responsible, at its sole cost and expense, for Marketing the Licensed Product in the Field in the Territory.
| 5.2 | Marketing Diligence |
Effective as of the Effective Date and during the term, HARROW shall use Commercially Reasonable Efforts to Commercialize the Licensed Product in the Field in the Territory for which the Marketing Authorization has been obtained. Without limiting the generality of the foregoing, HARROW shall:
| (a) | use Commercially Reasonable Efforts to maintain the U.S. MA, obtain and maintain the Marketing Authorization for Canada and obtain and maintain any other Regulatory Approval required for the Marketing of the Licensed Product in the Field in the Territory, including any required Pricing and Reimbursement Approval; |
| (b) | make the First Commercial Sale of the Licensed Product in the United States and Canada as soon as reasonably practicable following Regulatory Approval for such Licensed Product in the respective country; |
| (c) | use the ImprimisRx Pharmacy and customer service resources, seeking to transfer existing Klarity C prescriptions to the Licensed Product; |
| 19 |
| (d) | using HARROW’s sales organization, seeking to convert all current Klarity C prescribers to Licensed Product prescribers; |
| (e) | using HARROW market access resources, seeking commercial and non-commercial coverage for the Licensed Product; |
| (f) | actively Market the Licensed Product through trade shows, educating doctors of the Licensed Product’s unique characteristics and estimated availability; |
| (g) | actively Market the Licensed Product to HARROW’s database of customers and provide a telemedicine option to access the Licensed Product through HARROW’s visionology.com portal once the portal has been redesigned to accommodate the Licensed Product; |
| (h) | actively Market the Licensed Product to HARROW’s deep relationship in the United States laser eye surgery and laser vision correction (LASIK) market; |
| (i) | actively Market the Licensed Product to HARROW’s customer base of cataract surgeons; |
| (j) | perform marketing activities substantially in accordance with the Marketing Plan. |
| 5.3 | Marketing Plan and Reporting |
The initial U.S. MA marketing plan for the Licensed Product for the Territory is attached hereto as Exhibit 5.3 (“Marketing Plan”). Annually on or before October 1 of each year for the subsequent Calendar Year, HARROW shall make available to NOVALIQ, through the JSC, proposed updates and amendments to the Marketing Plan consistent with the level of detail in the Marketing Plan (including marketing budgets, marketing activities, timelines for such marketing activities, allocated resources, etc.) and the JSC will discuss and approve such proposed updates or amendments in the JSC. In addition, commencing on April 1 of the Calendar Year following the Calendar Year in which the First Commercial Sale in the United States has occurred, HARROW shall provide NOVALIQ on or before April 1 in each Calendar Year with a confidential summary of its Marketing activities performed in the Territory in the preceding year.
| 5.4 | Unauthorized Sales |
All Licensed Products Manufactured by or on behalf of HARROW shall be sold or otherwise distributed – and shall clearly indicate that they are – for use in the Field in the Territory only. HARROW shall contractually obligate the purchasers of Licensed Product to use Licensed Product, or actively resell or otherwise distribute Licensed Product in compliance with all Applicable Laws in the Territory governing the purchase, handling, sale and distribution of Licensed Products. HARROW shall use Commercially Reasonable Efforts to prevent the active sale (i.e., actively approaching individual customers) or other distribution of Licensed Product into, or for use in, countries outside the Territory or for use outside the Field. If HARROW becomes aware that a purchaser sells or otherwise distributes, or intends to sell or otherwise distribute, Licensed Product outside the Territory or outside the Field, it will promptly notify NOVALIQ in writing.
| 6. | Compliance |
In conducting their respective activities hereunder, each Party shall comply in all respects with all Applicable Laws, including but not limited to applicable export control, anti-bribery and corruption laws.
| 20 |
| 7. | Consideration |
| 7.1 | Upfront Payment |
In consideration for the rights and licenses granted under this Agreement, HARROW shall pay to NOVALIQ
| (a) | a one-time upfront payment in the amount of [***] upon the Signing Date (the “First Upfront Payment”). The First Upfront Payment shall be made by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW), with any transfer fees to be paid by HARROW, in immediately available funds within ten (10) Business Days following the Signing Date (invoicing to be made by way of a credit note); and |
| (b) | a one-time upfront payment in the amount of [***] upon the Effective Date (the “Second Upfront Payment”). The Second Upfront Payment shall be made by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW), with any transfer fees to be paid by HARROW, in immediately available funds within ten (10) Business Days following the Effective Date (invoicing to be made by way of a credit note). |
| 7.2 | Milestone Payments |
| (a) | Sales Milestones. |
HARROW shall make the following non-refundable, non-creditable sales milestone payments to NOVALIQ in connection with the first-time achievement of the applicable Milestone Event listed below (each, a “Milestone Event”), as set out below:
| # | Milestone Event | Payment (US$) | ||
| 1 | Aggregate annual Net Sales of Licensed Product in the Territory exceed [***] in a Calendar Year in the Territory | [***] in cash | ||
| 2 | Aggregate annual Net Sales of Licensed Product in the Territory exceed [***] in a Calendar Year in the Territory | [***] in cash | ||
| 3 | Aggregate annual Net Sales of Licensed Product in the Territory exceed [***] in a Calendar Year in the Territory | [***] in cash | ||
| 4 | Aggregate annual Net Sales of Licensed Product in the Territory exceed [***] in a Calendar Year in the Territory | [***] in cash |
HARROW shall notify NOVALIQ in writing of the achievement of any of the above sales-related Milestone Events by April 10th of the following Calendar Year (each, a “Milestone Notice”). For the avoidance of doubt, if more than one of the above Milestone Events occurs within one Calendar Year, HARROW shall make milestone payments for all such Milestone Events occurring in that specific Calendar Year.
| 21 |
| (b) | Payment Terms. Each cash milestone payment shall be paid by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW from time to time), with any transfer fees to be paid by HARROW, in immediately available funds within ten (10) Business Days following each Milestone Notice of a Milestone Event for which a Cash Payment is owed (invoicing to be made by way of a credit note). |
| (c) | Change of Control. If a Change of Control in HARROW or an Affiliate of HARROW holding rights to the NOVALIQ Technology (e.g. due to a sublicense by HARROW to such Affiliate or an assignment of this Agreement to such Affiliate) occurs at a time when the aggregate Net Sales of the Licensed Product in the Territory in the preceding twelve (12) months (or the entire period of time the Licensed Product has been sold in the Territory if it has been sold for less than twelve (12) months from the First Commercial Sale date) have [***], then HARROW shall pay to NOVALIQ, or shall procure that the surviving entity after the Change of Control pays to NOVALIQ, as applicable, an amount equal to ten percent (10%) of the Net Sales for such period (“Change of Control Payment”). HARROW shall notify NOVALIQ of the occurrence of a Change of Control event in HARROW within ten (10) Business Days following the closing of such transaction and the Change of Control Payment shall be due within twenty (20) Business Days thereafter. The Change of Control Payment shall not relieve HARROW, or the surviving entity after the Change of Control from the payment of future milestone or royalty payments; provided that HARROW, or the surviving entity after the Change of Control, as the case may be, may credit the Change of Control Payment against any future payments that will be owed to NOVALIQ under Sections 7.2, 7.3 and 7.4. |
| 7.3 | Minimum Annual License Fee |
Until the earlier of (a) the end of the Royalty Term, and (b) Harrow having achieved aggregate annual Net Sales of Licensed Product of [***]per Calendar Year in two (2) Calendar Years, HARROW shall pay to NOVALIQ a minimum annual license fee as set forth in the table below:
| Calendar Year | Amount of minimum annual license fee | |
| 2025 | [***] | |
| 2026 | [***] | |
| 2027 | [***] | |
| 2028 | [***] | |
| 2029 | [***] | |
| 2030 and thereafter | [***] |
| 22 |
HARROW shall pay the minimum annual license fee by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW from time to time), with any transfer fees to be paid by HARROW, in immediately available funds until January 31 of the Calendar Year for which the respective minimum annual license fee is due (invoicing to be made by way of a credit note).
| 7.4 | Royalties |
| (a) | Royalty Payments. During the Royalty Term HARROW shall pay to NOVALIQ royalties, calculated on the basis of aggregated annual Net Sales of Licensed Product in the Territory as follows: |
| Royalty Tiers | Royalty Rate | |
| For the portion of aggregated Net Sales of Licensed Product in the Territory that is lower than [***] | [***] | |
| For the portion of aggregated Net Sales of Licensed Product in the Territory that is equal to or exceeding [***] and below [***] | [***] | |
| For the portion of aggregated Net Sales of Licensed Product in the Territory that is equal to or exceeding [***] | [***] |
| (b) | Royalty Reduction. In any country of the Territory where the sale of the Licensed Product is not Covered by a Valid Patent Claim of the NOVALIQ Patent Rights, the royalty rates set forth in Sections 7.4(a) shall be reduced by [***]. HARROW may credit up to [***] of the amounts paid by HARROW to a Third Party, if any, for a license to any patent(s) that HARROW reasonably determines (in the opinion of independent patent counsel agreeable to both parties) would be infringed by the manufacturing, use or sale of the Licensed Product in the Field in the Territory against any royalties payable to NOVALIQ on and after the effective date of such Third Party license. However, under no circumstances shall the Royalty Rate paid by HARROW to NOVALIQ during the Royalty Term be less than [***] of the Royalty Rates set forth in Section 7.3. |
| (c) | Crediting of Minimum Annual License Fee. HARROW may credit the minimum annual license fee paid by HARROW to NOVALIQ under Section 7.3 in a Calendar Year against the royalty payments payable to NOVALIQ under Section 7.4 in such Calendar Year. |
By way of example: If in a given Calendar Year HARROW pays a minimum annual license fee of US$ 4,000,000 to NOVALIQ under Section 7.3, and NOVALIQ is entitled to receive royalties on Net Sales in the amount of US$ 9,000,000 in such Calendar Year under Section 7.4, then HARROW may credit the minimum annual license fee already paid to NOVALIQ against the royalties payable to NOVALIQ in such Calendar Year, and must pay royalties in the amount of US$ 5,000,000 in such Calendar Year.
| (d) | Conditions for Calculation. All royalty payments under this Section 7.4 are subject to the following conditions: |
| (i) | that only one royalty shall be due with respect to the same unit of Licensed Product; |
| 23 |
| (ii) | that no royalties shall be due upon the sale or other transfer among HARROW, its Affiliates and Sublicensees, but in such cases the royalty shall be due and calculated upon HARROW’s, its Affiliates’ or Sublicensees’ Net Sales to the first independent Third Party; |
| (iii) | that no royalties shall accrue on the sale or other disposition of Licensed Product by HARROW, its Affiliates or Sublicensees for use in a Clinical Trial; and |
| (iv) | that no royalties shall accrue on the disposition of Licensed Product in reasonable quantities by HARROW, its Affiliates or Sublicensees as samples (promotion or otherwise) or as donations (for example, to non-profit institutions or government agencies for a non-commercial purpose) if no Net Sales are generated by HARROW with the disposition of Licensed Product as samples or donations (and for the avoidance of doubt, HARROW may not deduct any costs in relation to such Licensed Product disposed as samples or donations from Net Sales), provided, however, that within any Calendar Year, commencing on or after the fourth year anniversary of the Effective Date of this Agreement, the free samples disposed of the Licensed Product shall not exceed five percent (5%) of the total quantities of Licensed Product sold. |
| (e) | Reports; Payment of Royalties. HARROW shall report to NOVALIQ the date of the First Commercial Sale of the Licensed Product within ten (10) Business Days of occurrence. In addition, during the term of this Agreement following the First Commercial Sale of the Licensed Product, HARROW shall furnish to NOVALIQ a written report for each Calendar Quarter showing the gross sales and the calculation of Net Sales (including the deductions from gross sales) of the Licensed Product by HARROW, its Affiliates and Sublicensees in the Territory during the reporting period and the royalties payable under this Agreement. Reports shall be due within forty five (45) calendar days after the end of each Calendar Quarter. At least once a year, HARROW’s independent auditors shall review royalty reports, calculations and associated payments to NOVALIQ. If any prior period adjustments are required by HARROW or its independent auditors to the Net Sales calculation, such adjustments shall be clearly noted in and applied to the following period royalty calculation, report and payment (e.g., the next quarterly report). Royalty payments shall be made by HARROW by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW from time to time), with any transfer fees to be paid by HARROW, in immediately available funds within five (5) Business Days after delivery of a final quarterly royalty report by way of a credit note. |
| 7.5 | Financial Audits |
| (a) | HARROW (including its Affiliates) and its Sublicensees shall keep complete and accurate books and records which may be necessary to ascertain properly and to verify the payments owed hereunder. Such records shall be open to inspection during usual business hours for a three (3) year period after the Calendar Year to which such records pertain. Once per Calendar Year, and no more than once for the records as to any given Calendar Year, NOVALIQ shall have the right to engage an independent Public Company Accounting Oversight Board (“PCAOB”) registered accounting firm reasonably acceptable to HARROW, at NOVALIQ’s expense, which shall have the right to examine in confidence the relevant HARROW records as may be reasonably necessary to determine and/or verify the amount of payments due hereunder. In the event there was an over-payment by HARROW hereunder, NOVALIQ shall promptly (but in no event later than thirty (30) calendar days after HARROW’s receipt of the independent auditor’s report) make payment to HARROW of any overpayment amounts. In the event that there was an under-payment by HARROW hereunder, HARROW shall promptly (but in no event later than thirty (30) calendar days after HARROW’s receipt of the independent auditor’s report) pay NOVALIQ the underpayment amount. In the event any payment by HARROW shall prove to have been incorrect by more than five percent (5%) to NOVALIQ’s detriment for the period audited, HARROW will pay the reasonable fees and costs of NOVALIQ’s independent auditor for conducting such audit. |
| 24 |
| (b) | If HARROW disputes the results of any audit conducted pursuant to Section 7.5(a), the Parties shall work in good faith to resolve the disagreement. If the Parties are unable to reach a mutually acceptable resolution of any such dispute within thirty (30) Business Days, the dispute shall be submitted for resolution to a certified PCAOB registered public accounting firm jointly selected by each Party’s certified public accountants or to such other person or entity as the Parties shall mutually agree (the “Auditor”). The decision of the Auditor shall be final and the costs of such procedure as well as the initial audit shall be borne between the Parties in such manner as the Auditor shall determine. If the Auditor determines that there has been an underpayment by HARROW, HARROW shall promptly pay to NOVALIQ the underpayment amount (but in no event later than fifteen (15) Business Days after the Auditor’s decision). If the Auditor determines that there has been an overpayment by HARROW, then NOVALIQ shall promptly reimburse HARROW the overpayment amount (but in no event later than fifteen (15) Business Days after the Auditor’s decision). |
| 7.6 | Taxes |
| (a) | Income Taxes. All payments under or in connection with this Agreement shall be inclusive of any income Taxes and each Party shall be responsible for its own income Taxes assessed by a Tax or other authority except as otherwise set forth in this Agreement. |
| (b) | Withholding Taxes. If any amount payable to NOVALIQ, its supplier or its licensee under this Agreement including without limitation under Sections 7.1, 7.2, 7.3 and 7.4(e) by HARROW is subject to withholding tax requirement under the applicable law of the Territory, the Parties agree to cooperate with one another and use reasonable efforts to reduce or eliminate Tax withholding or similar obligations in respect of any milestone, royalty or other payments paid by HARROW to NOVALIQ under this Agreement. Any Tax paid or required to be withheld by HARROW on account of any milestone, royalty or other payments payable to NOVALIQ under this Agreement may not be deducted from (and must be added to) the amount of payments to NOVALIQ. |
| (c) | VAT. All payments due to the terms of this Agreement are exclusive of value added Tax (VAT) or similar indirect Taxes. VAT and other similar indirect Taxes shall be borne by HARROW and added to the payments due under this Agreement. |
| 7.7 | Late Payments |
Any payment due to a payee from a payor that is past due under this Agreement shall bear interest in the amount of three percent (3%) per annum above SOFR, prorated to the actual days of delay.
| 25 |
| 8. | GOVERNANCE |
| 8.1 | Joint Steering Committee |
| (a) | JSC. The Parties agree to establish a Joint Steering Committee (“Joint Steering Committee” or “JSC”) to coordinate, oversee and monitor the further Development, Manufacture and Marketing of the Licensed Product in the Field in the Territory, and to facilitate the exchange of information between the Parties regarding the Development, Manufacture and Marketing of the Licensed Product in the Field in the Territory. |
| (b) | Establishment of the JSC. Promptly following the Effective Date, the Parties shall establish the JSC, to be composed of up to three (3) representatives of each Party, unless another number is agreed between the Parties. These representatives shall have appropriate technical credentials, experience and knowledge, and ongoing familiarity with the Licensed Product and sufficient seniority within the applicable Party consistent with the scope of the JSC’s responsibilities. A Party’s representatives will be appointed and may be replaced by such Party on written notice to the other Party, provided, however, that each Party will use reasonable efforts to ensure continuity of its representatives on the JSC. |
| (c) | Meetings. The JSC will coordinate and schedule JSC meetings, such meetings to take place at least once every Calendar Quarter, upon the reasonable request of either Party or as agreed between the Parties. JSC meetings may be conducted in person, by telephone or videoconference. The JSC will have a quorum if at least one (1) representative of each Party is present or participating. The Parties will endeavor to schedule meetings of the JSC at least two (2) months in advance. Each Party may invite guests to certain items on the agenda of the meetings, with reasonable prior notice, in order to discuss special technical or commercial topics. No guest shall have the right to vote at such meeting. In advance of any JSC meetings, the Parties will exchange copies of Development Data and other documents relating to the issues to be addressed at such meeting. The JSC shall be co-chaired by one representative of HARROW and one representative of NOVALIQ. The co-chairs shall have the responsibilities set forth in Section 8.1(d), but shall have no additional powers or rights beyond those held by the other JSC representatives. |
| (d) | Agendas and Meeting Minutes. The co-chairs shall be responsible for agreeing upon and distributing an agenda for each meeting of the JSC at least five (5) Business Days in advance of any such meeting. Each Party shall have the right to request the co-chairs to include any matter or issue related to the Development, Manufacture or Marketing of the Licensed Product on the agenda for a JSC meeting, which requests shall be accommodated by the co-chairs. The co-chairs shall alternate responsibility for generating and issuing minutes of each JSC meeting, which shall include a summary of any actions agreed at the meetings. The minutes will be issued in draft form and provided to the Alliance Managers and the JSC representatives of each Party for review. Any corrections or comments must be provided to the co-chair with responsibility for preparing the minutes within ten (10) Business Days after the draft minutes are issued, and such co-chair shall then issue the approved (or, if no comments are provided within such ten (10) Business Day period, deemed approved) minutes in final form to the Alliance Managers and the JSC representatives of each Party. |
| (e) | No Material Breach. For the avoidance of doubt, the Parties acknowledge and agree that a Party’s failure to satisfy the formal requirements set out in Section 8.1(c) and 8.1(d) or a failure of a Party to strictly comply with the terms set out in Section 8.1(c) and 8.1(d) will in principle not qualify as a material breach of this Agreement that entitles the other Party to terminate this Agreement pursuant to Section 14.3. |
| 26 |
| 8.2 | Functions and Powers of the JSC |
The JSC shall have strategic oversight over the Development, Manufacture and Marketing of the Licensed Product in the Field in the Territory, and shall be responsible for:
| (a) | overseeing the Development and Marketing of the Licensed Product; |
| (b) | discussing and approving updates and amendments to the Marketing Plan; |
| (c) | facilitating the exchange of information on the Development, Manufacturing and Marketing of the Licensed Product in accordance with this Agreement; and |
| (d) | such other responsibilities as may be assigned to the JSC pursuant to this Agreement or as may be mutually agreed upon by the Parties from time to time. |
The JSC shall have solely the powers expressly assigned to it in this Section 8 and elsewhere in this Agreement, and shall not have any power to amend, modify, or waive compliance with this Agreement.
| 8.3 | Decisions |
| (a) | Decision-making. The JSC will make decisions only by unanimous consent, with each Party having only one vote by its representatives (regardless of the number of each such representatives present or participating from a Party). |
| (b) | Final Decision-Making if Unanimous Consent is Not Obtained. If the JSC fails to reach unanimous agreement on a matter before it for decision for a period in excess of thirty (30) days, the matter may be referred by either Party to the Executive Officers, who shall meet in person or via teleconference within fourteen (14) Business Days and attempt to resolve such matter in good faith. If the Executive Officers fail to reach agreement as to such matter for a period in excess of ten (10) Business Days from their initial meeting, the final decision on such undecided matter shall be made by HARROW in good faith, provided that HARROW shall not: (i) unilaterally reduce its diligence obligations under this Agreement, including, but not limited to, its diligence obligations under Section 3, Section 4 and Section 5; or (ii) unilaterally amend the terms and conditions of this Agreement. |
| 8.4 | Alliance Managers |
| (a) | Appointment. Each Party shall appoint an employee who shall oversee interactions between the Parties for all matters related to this Agreement (each an “Alliance Manager”). The Alliance Managers shall have the right to attend all JSC meetings as non-voting participants and may bring to the attention of the JSC any matters or issues either of them reasonably believes should be discussed, and shall have such other responsibilities as set forth in Section 8.4(b) and elsewhere in this Agreement or as the Parties may mutually agree. Each Party may replace its Alliance Manager at any time or may designate different Alliance Managers by notice in writing to the other Party, provided, however, that each Party will use reasonable efforts to ensure continuity of its representatives on the JSC. |
| (b) | Responsibilities of the Alliance Managers. The Alliance Managers shall have the responsibility of creating and maintaining a constructive work environment between the Parties. Without limiting the generality of the foregoing, each Alliance Manager shall: |
| 27 |
| (i) | identify and bring issues that may result in disputes to the attention of the JSC in a timely manner, and function as the point of first referral in all matters of conflict resolution. In doing so, it is not intended that the Alliance Manager(s) act as a substitute for, or insert any delay in, the formal dispute resolution mechanisms set forth in this Agreement, but rather that the Alliance Manager(s) shall endeavor to maintain a positive and constructive relationship between the Parties at the working level; |
| (ii) | provide a single point of communication for seeking consensus both internally within the Parties’ respective organizations and between the Parties; |
| (iii) | plan and coordinate cooperative efforts, internal communications and external communications between the Parties with respect to this Agreement; and |
| (iv) | take responsibility for ensuring that meetings and the production of meeting agendas and minutes occur as set forth in this Agreement, and that relevant action items resulting from such meetings are appropriately carried out or otherwise addressed. |
| 9. | NOVALIQ RIGHT OF FIRST NEGOTIATION |
HARROW is currently developing a drug candidate for which an NDA may be submitted pursuant to Section 505(b)(2) of the Federal, Food, Drug and Cosmetic Act (“FFDCA”) to treat a rare form of cancer in the eye (the “HARROW Cancer Drug Candidate”). HARROW hereby grants to NOVALIQ, and NOVALIQ hereby accepts from HARROW, an exclusive right of first negotiation with respect to the HARROW Cancer Drug Candidate in accordance with this Section 9 (“ROFN”). If at any time during the Term, HARROW intends to out-license any rights, or otherwise grant any rights, to the HARROW Cancer Drug Candidate (“ROFN Transaction”), it shall promptly notify NOVALIQ thereof, and provide all information reasonably required by NOVALIQ for assessing its interest in such ROFN Transaction and be available to discuss such information. Within sixty (60) days upon receipt of such notification, including all reasonably required information (“ROFN Exercise Period”), NOVALIQ shall notify HARROW in writing whether it wishes to exercise its ROFN. If NOVALIQ does not exercise its ROFN within the ROFN Exercise Period, HARROW shall be free to negotiate an agreement for the ROFN Transaction with a Third Party. If NOVALIQ exercises its ROFN within the ROFN Exercise Period, the Parties shall initiate negotiations within thirty (30) days upon receipt of the ROFN exercise notice by HARROW, and shall negotiate exclusively and in good faith the terms of an agreement for the ROFN Transaction for a period of six (6) months, or such longer period as may be mutually agreed between the Parties (“ROFN Negotiation Period”). If the Parties fail to reach an agreement within the ROFN Negotiation Period, NOVALIQ’s rights under the ROFN Transaction shall expire and HARROW shall be free to negotiate an agreement for the ROFN Transaction with a Third Party.
| 10. | INTELLECTUAL PROPERTY |
| 10.1 | Inventorship; Ownership; Exploitation Rights |
| (a) | Inventorship. Inventorship of inventions conceived or reduced to practice during the course of the performance of activities pursuant to this Agreement shall be determined in accordance with U.S. patent laws. |
| (b) | Ownership. Subject to Section 10.2, as between the Parties, all Know-How and Inventions made, conceived or reduced to practice by a Party or any of its Affiliates’ employees, contractors or consultants in the course of conducting activities under this Agreement, together with all Patent Rights therein, shall be owned by such Party. All Know-How and Inventions made, conceived or reduced to practice jointly by each Party’s or any of its Affiliates’ employees, contractors or consultants in the course of conducting activities under this Agreement, together with all Patent Rights therein, shall be jointly owned by HARROW and NOVALIQ on the basis of an undivided interest and shall be deemed to be Controlled by each Party (the “Joint Know-How”, “Joint Inventions”, and “Joint Patent Rights”, as applicable, and together the “Joint IP”); provided that each Party may only use the Joint IP as provided for in Section 10.1(c). Each Party hereby authorizes and grants the other Party its permission and consent to assume, directly or through its authorized agents, attorneys, or representatives, the responsibilities, and to exercise the rights as set forth in Sections 10.2,10.3,10.4 and 10.5. |
| 28 |
| (c) | Exploitation Rights. Subject to the terms and conditions of this Agreement, (i) HARROW shall have the sole and exclusive right to use and exploit (which shall include the right to grant licenses in multiple tiers) the Joint IP and Improvements (including Improvement Patent Rights) for the Development, Manufacture and Marketing of the Licensed Products in the Field in the Territory, and (ii) NOVALIQ shall have the sole and exclusive right to use and exploit (which shall include the right to grant licenses in multiple tiers) the Joint IP and Improvements (including Improvement Patent Rights) outside the Field in the Territory, and inside and outside the Field in the ROW, and for other products without any compensation to HARROW; in each case of (i) and (ii) in accordance with the terms and conditions of this Agreement |
| (d) | Assignment of Intellectual Property Rights by Subcontractors. To the extent that a Party utilizes contractors or consultants to perform any of its activities under this Agreement, such Party shall ensure that any such contractor or consultant is obligated to assign all of its rights, title and interests to any Inventions, Patent Rights and/or Know-How made by such contractor or consultant in the performance of such activities to the Party engaging such contractor or consultant to give effect to the ownership provisions and exploitation rights laid down in this Section 10.1. |
| 10.2 | Filing, Prosecution and Maintenance of Patent Rights |
| (a) | NOVALIQ Patent Rights. NOVALIQ shall be responsible for filing, prosecuting and maintaining the NOVALIQ Patent Rights (other than Joint Patent Rights), including any post-grant proceedings in the Territory at NOVALIQ’s expense. Notwithstanding the above, in the event that a post-grant proceeding in the Territory is filed in response to litigation brought by HARROW pursuant to Section 10.3, the Parties agree to work together to make sure that the legal positions taken in the litigation and the post-grant proceedings are aligned. |
| (b) | Joint Patent Rights in the Territory. HARROW shall be responsible to file, prosecute, and maintain (i) the Joint Patent Rights in the Territory in the name of both Parties and (ii) the Improvement Patent Rights in the Territory at HARROW’s name, in each case of (i) and (ii) at HARROW’s expense. If HARROW declines to file, prosecute, or maintain any Joint Patent Right or an Improvement Patent Right in any country of the Licensed Territory, or desires to allow any Joint Patent Right or Improvement Patent Right to lapse in any country of the Licensed Territory, then: |
| (i) | HARROW shall provide NOVALIQ with reasonable written notice of such decision so as to permit NOVALIQ to decide whether to file, prosecute, or maintain such Joint Patent Right or Improvement Patent Rights in the Territory in its own name and to take any necessary action. |
| 29 |
| (ii) | Following notice from HARROW pursuant to Section 10.2(b)(ii), NOVALIQ may, by providing prompt written notice thereof to HARROW, assume control of the filing, prosecution or maintenance of such Joint Patent Right or Improvement Patent Right in its own name, and ownership of such Joint Patent Right or Improvement Patent Right shall be fully transferred to NOVALIQ, all at NOVALIQ’s expense, and HARROW’s right to use such Joint Patent Right or Improvement Patent Right shall cease. |
| (c) | Joint Patent Rights in the ROW. NOVALIQ shall have the right to file, prosecute, and maintain the Joint Patent Rights and Improvement Patent Rights in its own name in the ROW at NOVALIQ’s expense, and such Joint Patent Rights and Improvement Patent Rights outside the Territory shall be solely owned by NOVALIQ. Notwithstanding the above, in the event that common ownership of the Joint Patent Rights or Improvement Patent Rights is required to maximize protection either in or outside of the Territory, the Parties will work together to establish common ownership of such Joint Patent Rights or Improvement Patent Rights, provided that exploiting such rights shall be in accordance with Section 10.1(c). |
| (d) | Orange Book Listing, Patent Term Extensions. NOVALIQ shall have sole decision-making authority, at its expense, to select the NOVALIQ Patent Rights and Joint Patent Rights for which (i) listing in the publication, Approved Drug Products With Therapeutic Equivalence Evaluations published by the FDA in the United States ( commonly known as the “Orange Book”) or any analogous or similar listing in the Territory, and/or (ii) a patent term extension is to be sought or obtained with respect to the Licensed Product in the Territory; provided that the Parties shall consult with and cooperate and coordinate with each other through the JSC, including by providing necessary information and assistance. Notwithstanding the above, NOVALIQ shall not remove or delist any patents from the Orange Book listing for the Licensed Product unless agreed to by both Parties or as is required by law. |
| (e) | Cooperation. The Parties shall cooperate and consult with each other with respect to the filing, prosecution and maintenance of Joint Patent Rights and Improvement Patent Rights inside and outside the Territory, including with respect to terminal disclaimers, obviousness type double patenting and allowance. |
| 10.3 | Enforcement of NOVALIQ Patent Rights and of Joint Patent Rights |
| (a) | Notice. Each Party shall promptly provide the other Party with written notice reasonably detailing any known or alleged infringement by a Third Party of any NOVALIQ Patent Right or Joint Patent Right affecting the Licensed Product in the Field in the Territory (“Third Party Infringement”). |
| (b) | Enforcement. |
| (i) | With respect to the NOVALIQ Product Patent Rights and the Joint Patent Rights, HARROW shall have the right to determine and control a course of action designed to curtail any Third Party Infringement in the Field in the Territory at its own expense. HARROW shall keep NOVALIQ reasonably informed as to any legal courses of action it pursues pursuant to this Section 10.3(b). If such course of action includes litigation, to the extent permitted by Applicable Law, NOVALIQ shall have the right to be represented in such litigation by independent counsel at its own expense. At the request and at the cost of HARROW, NOVALIQ shall provide reasonable assistance to HARROW in connection with such enforcement, including by executing reasonably appropriate documents, cooperating in discovery, and joining as a party to the action. |
| 30 |
| (ii) | If HARROW elects not to enforce its rights under a NOVALIQ Product Patent Right or a Joint Patent Right against such Third Party Infringement or not to further pursue the enforcement of its rights, it shall promptly notify NOVALIQ thereof. In such case, NOVALIQ shall have the right to determine and control the course of action designed to curtail such Third Party Infringement at its own expense. NOVALIQ shall keep HARROW reasonably informed as to any legal courses of action it pursues pursuant to this Section 10.3(b). At the request of NOVALIQ, HARROW shall provide reasonable assistance to NOVALIQ in connection with such enforcement, including by executing reasonably appropriate documents, cooperating in discovery, and joining as a party to the action. |
| (iii) | With regard to the NOVALIQ Platform Patent Rights, NOVALIQ shall have the right to determine and control a course of action designed to curtail any Third Party Infringement in the Field in the Territory at its own expense. NOVALIQ shall keep HARROW reasonably informed as to any legal courses of action it pursues pursuant to this Section 10.3(b). At the request and at the cost of NOVALIQ, HARROW shall provide reasonable assistance to NOVALIQ in connection with such enforcement, including by executing reasonably appropriate documents, cooperating in discovery, and joining as a party to the action. Notwithstanding the above, in the event that, with respect to the Licensed Product, a Third Party provides notice to NOVALIQ or HARROW pursuant to 21 CFR 314.52 or 21 CFR 314.95 with respect to any NOVALIQ Platform Patent Rights, NOVALIQ shall advise HARROW within thirty (30) days of its receipt of such notice whether it intends to file an infringement suit against such Third Party. If NOVALIQ advises HARROW that it does not intend to file suit, HARROW shall then be granted the right to file suit against such Third Party on or before forty five (45) days from when HARROW or NOVALIQ received notice from the Third Party, and such NOVALIQ Platform Patent Rights shall be treated as if they were NOVALIQ Product Patent Rights for purposes of such suit and this Article 10. |
| (iv) | Neither Party shall settle any dispute in connection with a Third Party Infringement of any NOVALIQ Product Patent Right, Improvement Patent Right or Joint Patent Right in the Field in the Territory without the other Party’s prior written approval, such approval not to be unreasonably conditioned, withheld or delayed. Notwithstanding the above, in any Third Party Infringement that results in a so called Paragraph IV Hatch Waxman litigation in the US and wherein the Licensed Product is the Reference Listed Drug, HARROW shall have the sole authority to settle such action without NOVALIQ’s consent, provided that the terms of such settlement do not negatively impact the scope of the claims of the NOVALIQ Product Patent Rights, or the validity or enforceability of the NOVALIQ Product Patent Rights, or place any obligation or liability on NOVALIQ, and further provided that the scope of any such settlement is strictly limited to the product for which the Third Party submitted its Abbreviated New Drug Application or New Drug Application referencing the Licensed Product. For the avoidance of doubt, and except as set forth in the last two sentences of Section 10.3(b)(iii), HARROW shall not have the authority to settle any Infringement of a NOVALIQ Platform Patent Right, and NOVALIQ shall have the sole right and authority to settle any Infringement of a NOVALIQ Platform Patent Right, but if the Infringement of the NOVALIQ Platform Patent Right is a so called Paragraph IV Hatch Waxman litigation in the United States wherein the Licensed Product is the Reference Listed Drug, NOVALIQ shall obtain HARROW’s prior consent before entering into any settlement. |
| 31 |
| (v) | Any recoveries resulting from such an action relating to a claim of Third Party Infringement shall be first applied to reimburse the Parties’ costs in connection with the Third Party Infringement, and the remaining portion shall be retained by the Party who enforced the NOVALIQ Patent Right, Improvement Patent Right or Joint Patent Right in the Field in the Territory, with any amounts recovered by HARROW, if HARROW is the enforcing Party, being treated as Net Sales of HARROW for calculating sales milestone payments and royalties under Section 7.2 and Section 7.4. |
| (c) | Patent Marking. HARROW, its Affiliates and Sublicensees: (a) may mark the Licensed Product with the number of each issued patent under the NOVALIQ Patent Rights that applies to the Licensed Product; and (b) shall comply with the patent marking statutes in each country in which the Licensed Product is Manufactured by or on behalf of HARROW, its Affiliates and Sublicensees. |
| 10.4 | Defense of Third Party Infringement Claims |
| (a) | Notice. If a Party becomes aware of any actual or potential claim alleging that the Development, Manufacture, or Commercialization of the Licensed Products in the Field in the Territory infringes, misappropriates, or otherwise violates any intellectual property rights of a Third Party (or would if carried out) (“Third Party Allegation”), then such Party will notify the other Party as promptly as possible following knowledge of such potential claim, or the receipt of service of process in such action, suit, or proceeding, or the date on which such Party becomes aware that such action, suit, or proceeding has been instituted, and the JSC will meet as soon as possible to discuss and determine the overall strategy for defense of such matter. |
| (b) | Defense. |
| (i) | HARROW shall have the primary right to determine and control a course of action designed to defend against Third Party Allegations in the Field in the Territory at its own expense. HARROW shall keep NOVALIQ reasonably informed with respect to the defense of such Third Party Allegations. NOVALIQ shall have the right to be represented in such litigation by independent counsel at its own expense. At the request and cost of HARROW, NOVALIQ shall provide reasonable assistance to HARROW in connection with such defense, including by executing reasonably appropriate documents, cooperating in discovery, and joining as a party to the action. |
| (ii) | If HARROW elects not to defend against any such Third Party Allegations, it shall promptly notify NOVALIQ thereof. In such case, NOVALIQ shall have the right to determine and control the course of action at its own expense. NOVALIQ shall keep HARROW reasonably informed with respect to the defense of such Third Party Allegations. HARROW shall have the right to be represented in such litigation by independent counsel at its own expense. At the request of NOVALIQ, HARROW shall provide reasonable assistance to NOVALIQ in connection with such defense, including by executing reasonably appropriate documents, cooperating in discovery, and joining as a party to the action. |
| 32 |
| (iii) | Neither Party shall settle any dispute in connection with Third Party Allegations without the other Party’s prior written approval, such approval not to be unreasonably conditioned, withheld or delayed. |
| 10.5 | Joint Patent Rights and Improvement Patent Rights outside the Territory |
NOVALIQ shall have the sole right to enforce and defend the Joint Patent Rights and the Improvement Patent Rights outside the Territory at its cost.
| 10.6 | NOVALIQ Trademarks |
HARROW shall use the NOVALIQ Trademarks (i.e., VEVYE and EYESOL), as needed, in the United States to mark the Licensed Product. NOVALIQ shall have the sole right to file, prosecute, maintain, defend and enforce the NOVALIQ Trademarks in the Territory.
| 10.7 | VEVYE Domain |
NOVALIQ shall remain the owner of the domain www.veyve.com, but NOVALIQ shall grant HARROW the exclusive right to control the management of any website published on the subject domain, and shall transfer the domain to HARROW’s web hosting provider for the publication of content. HARROW shall be responsible for creating any content that is specifically related to the Licensed Product in the Field in the Territory, NOVALIQ will be responsible for creating all other content. HARROW covenants to publish NOVALIQ’s content for the Licensed Product outside the Field or outside the Territory.
| 11. | Confidentiality |
| 11.1 | Obligation of Confidentiality |
As of and after the Signing Date, all Confidential Information disclosed, revealed or otherwise made available to one Party (“Receiving Party”) by or on behalf of the other Party (“Disclosing Party”) under, or as a result of, this Agreement is made available to the Receiving Party solely to permit the Receiving Party to exercise its rights, and perform its obligations, under this Agreement. The Receiving Party shall not use any of the Disclosing Party’s Confidential Information for any other purpose, and shall not disclose, reveal or otherwise make any of the Disclosing Party’s Confidential Information available to any other person, firm, corporation or other entity, without the prior written authorization of the Disclosing Party, except as explicitly stated in this Section 11. In addition, as of the Signing Date, the Confidentiality Agreement shall terminate and its terms shall be replaced by this Agreement, and more specifically this Article 11.
| 11.2 | Additional Obligations |
| (a) | Appropriate Safeguards. In furtherance of the Receiving Party’s obligations under Section 11.1 hereof, the Receiving Party shall take all reasonable steps, and shall implement all appropriate and reasonable safeguards, to seek to prevent the unauthorized use or disclosure of any of the Disclosing Party’s Confidential Information. |
| (b) | Unauthorized Use or Disclosure. The Receiving Party shall furnish the Disclosing Party with written notice immediately of it becoming aware of any unauthorized use or disclosure of any of the Disclosing Party’s Confidential Information by any officer, employee, assignee, licensee or Sublicensee, or potential assignee, contract service provider, and its licensee or Sublicensee, or consultant, investor or potential investor of the Receiving Party, and shall take all actions reasonably required in order to prevent any further unauthorized use or disclosure of the Disclosing Party’s Confidential Information. |
| 33 |
| 11.3 | Limitations |
The Receiving Party’s obligations under Sections 11.1 and 11.2 shall not apply to the extent that the Receiving Party can demonstrate by competent evidence that any of the Disclosing Party’s Confidential Information:
| (a) | is known by the Receiving Party at the time of its receipt, and not through a prior disclosure by the Disclosing Party, as documented by the Receiving Party’s business records; |
| (b) | is in the public domain by use and/or publication before its receipt from the Disclosing Party, or thereafter enters the public domain through no fault of the Receiving Party; |
| (c) | is subsequently disclosed to the Receiving Party by a Third Party who may lawfully do so and is not under an obligation of confidentiality to the Disclosing Party; |
| (d) | is developed by the Receiving Party independently of Confidential Information received from the Disclosing Party, as documented by the Receiving Party’s business records; or |
| (e) | is disclosed to judicial, governmental or other regulatory agencies in order to obtain patents or to gain or maintain approval to conduct Clinical Trials or to Market Licensed Product, but such disclosure may be only to the extent reasonably necessary to obtain patents or authorizations and is subject to Section 11.4(b). |
| 11.4 | Authorized Disclosures |
| (a) | Necessary Disclosures. Each Party may disclose the other Party’s Confidential Information to the extent reasonably: |
| (i) | deemed necessary by the Receiving Party to be disclosed to such Party’s Affiliates, the Receiving Party’s Sublicensees, subcontractors, agent(s), consultant(s), and/or other Third Parties for the Development, Manufacturing and/or Marketing of the Licensed Product (or for such entities to determine their interest in performing such activities) in accordance with this Agreement on the condition that such Affiliates or Third Parties agree to be bound by confidentiality and non-use obligations that substantially are no less stringent than those confidentiality and non-use provisions contained in this Agreement; |
| (ii) | in the case of NOVALIQ, to its shareholders on the condition that such shareholders are bound by confidentiality and non-use obligations that substantially are no less stringent than those confidentiality and non-use provisions contained in this Agreement; |
| (iii) | deemed necessary by counsel to the Receiving Party to be disclosed to such Party’s attorneys, independent accountants or financial advisors for the sole purpose of enabling such attorneys, independent accountants or financial advisors to provide advice to the Receiving Party, on the condition that such attorneys, independent accountants and financial advisors agree to be bound by the confidentiality and non-use obligations contained in this Agreement; or |
| (iv) | deemed necessary by the Receiving Party to be disclosed to any bona fide potential or actual licensee, sublicensee, acquirer, merger partner, investor, investment banker, bank, rating agency, insurer, or other potential or actual partner, collaborator or funding source; provided that in connection with such disclosure, the Disclosing Party shall inform each disclosee of the confidential nature of such Confidential Information and disclosure shall be subject the agreement of each disclosee to be bound by confidentiality and non-use obligations that substantially are no less stringent than those confidentiality and non-use provisions contained in this Agreement. |
| 34 |
| (b) | Required Disclosures. If a Party is required by judicial, governmental or administrative process to disclose Confidential Information that is subject to the non-disclosure provisions of Section 11.1, such Party shall promptly inform the other Party of the disclosure that is being sought in order to provide the other Party an opportunity to challenge or limit the disclosure obligations. Confidential Information that is disclosed by judicial or administrative process shall remain otherwise subject to the confidentiality and non-use provisions of this Section 11, and the Party disclosing Confidential Information pursuant to law or court order shall take all steps reasonably necessary, including without limitation obtaining an order of confidentiality, to ensure the continued confidential treatment of such Confidential Information. |
| 11.5 | Survival |
All of the Receiving Party’s obligations under this Section 11 hereof, with respect to the protection of the Disclosing Party’s Confidential Information, shall survive the expiration or termination of this Agreement for a period of ten (10) years.
| 11.6 | Public Announcements, Press Releases |
The Parties agree that the material terms of this Agreement are the Confidential Information of both Parties, subject to the special authorized disclosure provisions set forth in Sections 11.2 and 11.3 and this Section 11.6. The Parties have agreed to make a joint press release of the execution of this Agreement and will agree on the wording of such joint press release following the Effective Date. After the release of such joint press release, no disclosure of the existence, or the terms, of this Agreement may be made by either Party without first obtaining the prior written approval of the other Party and agreement upon the nature and text of such public announcement and no Party shall use the name, trademark, trade name or logo of the other Party, its Affiliates or their respective employee(s) in any publicity, promotion, news release or disclosure relating to this Agreement or its subject matter, without the prior express written permission of the other Party, except as may be required by Applicable Law or as otherwise expressly provided in this Agreement. Notwithstanding the aforementioned, NOVALIQ is entitled to use the name and logo of the Licensed Product and HARROW for public relation purposes in and outside of the Territory. The Parties agree that NOVALIQ shall be entitled to make a press release upon the achievement of each milestone and each milestone trigger of a royalty payment under this Agreement and HARROW shall refuse its permission to such press release only for cause and by suggesting amendments to the press release which would allow HARROW to grant its permission. No permission shall be required to the extent a Party is legally required (i) by Applicable Laws; (ii) by the listing standards or agreements of any national or international securities exchange or similar laws of a governmental authority, market or agency; or (iii) in a judicial, administrative or arbitration proceedings, provided that in such instances the other Party shall be entitled to make a corresponding public announcement.
| 11.7 | Publications |
HARROW and NOVALIQ each acknowledge the other Party’s interest in publishing the results of its research in order to obtain recognition within the scientific community and to advance the state of scientific knowledge. Each Party also recognizes the mutual interest in obtaining valid patent protection and in protecting business interests and trade secret information. Consequently, except for disclosures permitted pursuant to Section 11.4, HARROW, its employee(s) or consultant(s) wishing to make a publication covering its activities under this Agreement shall deliver to NOVALIQ a copy of the proposed written publication or an outline of an oral disclosure at least forty-five (45) calendar days prior to submission for publication or presentation. NOVALIQ shall have the right (a) to propose modifications to the publication or presentation for patent reasons, trade secret reasons or business reasons or (b) to request a reasonable delay in publication or presentation in order to protect patentable information. If NOVALIQ requests a delay, HARROW shall delay submission or presentation for a period of sixty (60) calendar days to enable patent applications protecting each Party’s rights in such information to be filed in accordance with Section 10.2. Upon expiration of such sixty (60) calendar days, HARROW shall be free to proceed with the publication or presentation. If NOVALIQ requests modifications to the publication or presentation, HARROW shall edit such publication to prevent disclosure of trade secret or proprietary business information prior to submission of the publication or presentation. Each Party’s contribution shall be acknowledged in any publication by co-authorship or other acknowledgement, whichever is appropriate in accordance with customary scientific practice. For the avoidance of doubt, once written approval has been granted for a particular disclosure, such disclosed information may be subsequently disclosed without the requirement of further approvals. NOVALIQ shall use reasonable efforts not to publish or publicly disclose research results that could negatively impact the Development or Marketing of the Product in the Field in the Territory.
| 35 |
| 12. | Warranties and Limitation of Liability |
| 12.1 | Representations and Warranties of each Party |
Each of NOVALIQ and HARROW hereby represents and warrants to the other Party hereto as follows as of the Effective Date:
| (a) | it is a corporation or entity duly organized and validly existing under the laws of the state or other jurisdiction of its incorporation or formation; |
| (b) | the execution, delivery and performance of this Agreement by such Party does not conflict with any other agreement by which it is bound, and has been duly authorized by all requisite corporate action and does not require any shareholder action or approval |
| (c) | it has the power and authority to execute and deliver this Agreement and to perform its obligations hereunder; |
| (d) | this Agreement is a legal and valid obligation binding upon such Party and enforceable in accordance with its terms, subject to the effects of bankruptcy, insolvency, or other laws of general application affecting the enforcement of creditor rights, judicial principles affecting the availability of specific performance, and general principles of equity (whether enforceability is considered a proceeding at law or equity); |
| (e) | it is not aware of any government authorization, consent, approval, license, exemption of or filing or registration with any court or governmental department, commission, board, bureau, agency or instrumentality, domestic or foreign, under any Applicable Laws, currently in effect, necessary for, or in connection with, the transactions contemplated by this Agreement or any other agreement or instrument executed in connection herewith, or for the performance by it of its obligations under this Agreement; and |
| (f) | neither it, nor any of its respective Affiliates has been debarred by the FDA, is not subject to any similar sanction of other Regulatory Authorities in the Territory, and, to its Knowledge, neither Party nor any of its respective Affiliates has used, or will engage, in any capacity, in connection with this Agreement or any ancillary agreements (if any), any Person who either has been debarred by any Regulatory Authority, or is the subject of a conviction described in Section 306 of the FFDCA. |
| 36 |
| 12.2 | Additional Representations and Warranties of NOVALIQ |
NOVALIQ represents and warrants to HARROW that:
| (a) | to NOVALIQ’s Knowledge, the NOVALIQ Patent Rights in existence as of the Signing Date exist and are not invalid or unenforceable, in whole or in part; |
| (b) | as of the Effective Date, it has the full right, power and authority to grant the license under Section 2.1; |
| (c) | it has not previously assigned, transferred, conveyed or otherwise encumbered its right, title and interest in the NOVALIQ Patent Rights or NOVALIQ Know-How affecting the Development, Manufacture and/or Marketing of the Licensed Product in the Territory and in the Field by HARROW after the Effective Date. More specifically, NOVALIQ warrants as of the Effective Date that it has not granted to any Third Party rights, in the Territory and in the Field for the combination of: (i) a calcineurin inhibitor as the active ingredient; and (ii) either: (a) the inactive delivery vehicle that is currently used in the Licensed Product; or (b) the delivery vehicle in the NOV03 Product (i.e. perfluorohexyloctane); |
| (d) | to NOVALIQ’s Knowledge at the Signing Date, it is the sole and exclusive owner of the NOVALIQ Patent Rights and the NOVALIQ Know-How, all of which are free and clear of any liens, charges and encumbrances affecting the Development, Manufacture and/or Marketing of the Licensed Product in the Territory and in the Field, and, to NOVALIQ’s Knowledge at the Signing Date, no other person, corporate or other private entity, or governmental entity or subdivision thereof, has any claim of ownership whatsoever with respect to the NOVALIQ Patent Rights and NOVALIQ Know-How; |
| (e) | to NOVALIQ’s Knowledge at the Signing Date, the exercise of the license granted to HARROW under the NOVALIQ Patent Rights and the NOVALIQ Know-How as of the Effective Date, including without limitation the Development, Manufacture and Marketing of the Licensed Product by HARROW as of the Effective Date does not infringe any intellectual property rights owned or possessed by any Third Party; |
| (f) | to NOVALIQ’s Knowledge at the Signing Date, there are no claims, judgments or settlements against or owed by NOVALIQ and no pending or threatened claims or litigation relating to the NOVALIQ Patent Rights and NOVALIQ Know-How; |
| (g) | to NOVALIQ’s Knowledge at the Signing Date, the development of the Licensed Product until the Signing Date has been conducted in conformance with cGMP and all Applicable Laws; and |
| (h) | other than the Licensed Product and the NOV03 Product, NOVALIQ is not as of the Signing Date developing a product in the Field intended for marketing in the Territory. |
| 12.3 | Additional Representations and Warranties of HARROW |
| 12.4 | Covenants of NOVALIQ |
| (a) | During the Term, NOVALIQ shall not, and shall cause its Affiliates not to, grant to any Third Party rights that conflict with the rights granted to Harrow for the Field for the Territory, or otherwise assign, transfer, license, convey, or otherwise encumber its right, title, or interest in or to the NOVALIQ Technology, the Joint Patent Rights and Joint Know-How or the Licensed Product for the Field for the Territory. More specifically, NOVALIQ covenants that neither it, not its Affiliates, will grant to any Third Party rights, in the Territory and in the Field for the combination of: (i) a calcineurin inhibitor as the active ingredient; and (ii) either: (a) the inactive delivery vehicle that is currently used in the Licensed Product; or (b) the delivery vehicle in the NOV03 Product (i.e. perfluorohexyloctane). For the avoidance of doubt, NOVALIQ shall not be restricted in any way with respect to the Licensed Product outside the Territory or inside the Territory outside the Field. |
| 37 |
| (b) | To the extent permissible under Applicable Laws all employees, agents, advisors, consultants or contractors of NOVALIQ shall be under an obligation to assign all of their right, title and interest in and to any NOVALIQ Technology and any Joint Technology, whether or not patentable, and intellectual property rights therein, to NOVALIQ as the owner thereof. HARROW shall have no obligation to contribute to any remuneration of any inventor employed or previously employed by NOVALIQ in respect of any inventions, information and other discoveries made in furtherance of this Agreement, including any intellectual property rights therein, which are assigned to NOVALIQ. NOVALIQ shall pay all such remuneration due to such inventors with respect to such inventions. |
| 12.5 | Covenant of HARROW. To the extent permissible under Applicable Laws all employees, agents, advisors, consultants or contractors of HARROW shall be under an obligation to assign all of their right, title and interest in and to any Improvements and any Joint Technology, whether or not patentable, and intellectual property rights therein, to HARROW as the owner thereof. NOVALIQ shall have no obligation to contribute to any remuneration of any inventor employed or previously employed by HARROW in respect of any inventions, information and other discoveries made in furtherance of this Agreement, including any intellectual property rights therein, which are assigned to HARROW. HARROW shall pay all such remuneration due to such inventors with respect to such inventions. |
| 12.6 | Disclaimer of Warranty |
Except as expressly set forth in this Agreement, each Party expressly disclaims, waives, releases, and renounces any representation or warranty of any kind, express or implied either in fact or by operation of law, by statute or otherwise, whether written or oral, or arising from course of performance, course of dealing or usage of trade, including, without limitation, any representation or warranty with respect to non-infringement, value, adequacy, freedom from fault, quality, efficiency, suitability, characteristics or usefulness, or merchantability or fitness for a particular purpose. WITHOUT LIMITING THE GENERALITY OF THE FOREGOING, HARROW ACKNOWLEDGES AND AGREES THAT NOVALIQ HAS NOT PERFORMED A FREEDOM TO OPERATE ANALYSIS FOR THE LICENSED PRODUCT FOR THE TERRITORY.
| 12.7 | Limitation of Liability |
EXCEPT IN CASE OF THIRD PARTY INDEMNIFICATION under Section 13, A breach of confidentiality under Section 11 or a breach of the non-compete under Section 2.4 and Except in the case of willful or intentional misconduct or gross negligence, neither Party shall be liable to the other Party for any indirect, punitive or consequential damages, or for damages for loss of profits or loss of business opportunity, whether based on contract or tort, or arising under Applicable Laws or otherwise.
| 38 |
| 13. | Indemnification |
| 13.1 | NOVALIQ’s Obligations to Indemnify |
NOVALIQ shall indemnify, defend and hold HARROW, its Affiliates and Sublicensees and their respective employees, agents, officers, and directors (individually and/or collectively referred to hereinafter as a “HARROW Indemnitee”) harmless from and against any and all Third Party claims, suits, actions, demands, liabilities, expenses and/or loss, including reasonable legal expense and attorneys’ fees (collectively, “Losses”) to the extent such Losses arise directly or indirectly out of a claim, suit or other proceeding made or brought by a Third Party against HARROW or a HARROW Indemnitee (a “HARROW Claim”) based on, resulting from, or arising in connection with:
| (a) | any breach of any of NOVALIQ’s representations, warranties or covenants set forth in this Agreement, including Section 12.2(c) and Section 12.4(a); |
| (b) | any grossly negligent, willful or intentional misconduct, error or omission on the part of NOVALIQ or any NOVALIQ Indemnitee; or |
| (c) | NOVALIQ’s or NOVALIQ Indemnitee’s failure to comply with Applicable Laws when performing its obligations under this Agreement; |
provided, however, that NOVALIQ shall not be obligated to indemnify, defend or hold harmless HARROW or a HARROW Indemnitee from any HARROW Claim or for any Losses incurred by HARROW or a HARROW Indemnitee: (i) to the extent arising out of, or attributable to a breach by HARROW, or any HARROW Indemnitee of any representation or warranty of HARROW, or a HARROW Indemnitee contained in this Agreement; (ii) to the extent arising out of, or attributable to HARROW’s or HARROW Indemnitee’s failure to comply with Applicable Laws in connection with this Agreement; (iii) to the extent arising out of, or attributable to any grossly negligent, willful or intentional misconduct, error or omission on the part of HARROW or a HARROW Indemnitee; or (iv) otherwise are Losses as to which HARROW indemnifies NOVALIQ as set forth in Section 13.2.
| 13.2 | HARROW’s Obligations to Indemnify |
HARROW shall indemnify, defend and hold NOVALIQ, its Affiliates and their respective employees, agents, officers, and directors (individually and/or collectively referred to hereinafter as a “NOVALIQ Indemnitee”) harmless from and against any and all Losses to the extent such Losses arise directly or indirectly out of a claim, suit or other proceeding made or brought by a Third Party against NOVALIQ or an NOVALIQ Indemnitee (an “NOVALIQ Claim”) based on, resulting from, or arising in connection with:
| (a) | any breach of any of HARROW’s representations, warranties or covenants set forth in this Agreement; |
| (b) | any grossly negligent, willful or intentionally wrongful act, error or omission on the part of HARROW or any HARROW Indemnitee; |
| (c) | HARROW’s or HARROW Indemnitee’s failure to comply with Applicable Laws in connection with this Agreement; or |
| (d) | the Development, Manufacture, Marketing or other use of the Licensed Product by HARROW; |
provided, however, that HARROW shall not be obligated to indemnify, defend or hold harmless NOVALIQ or an NOVALIQ Indemnitee from any NOVALIQ Claim or for any Losses incurred by NOVALIQ or an NOVALIQ Indemnitee: (i) to the extent they are arising out of or attributable to a breach by NOVALIQ, or any NOVALIQ Indemnitee of any representation or warranty of NOVALIQ, or an NOVALIQ Indemnitee contained in this Agreement; (ii) to the extent arising out of, or attributable to any grossly negligent, willful or intentional misconduct, error or omission on the part of NOVALIQ, or an NOVALIQ Indemnitee; or (iii) otherwise are Losses as to which NOVALIQ indemnifies HARROW as set forth in Section 13.1.
| 39 |
| 13.3 | Indemnification Procedures. |
| (a) | Procedure. Each indemnified Party shall notify in English the indemnifying Party in writing (and in reasonable detail) of the claim within ten (10) Business Days after receipt by such indemnified Party of notice of the HARROW Claim or NOVALIQ Claim, as the case may be, or otherwise becoming aware of the existence or threatened existence thereof (such HARROW Claim or NOVALIQ Claim being referred to as a “Claim”). Failure to give such notice shall not constitute a defense, in whole or in part, to any Claim by an indemnified Party hereunder except to the extent the rights of the indemnifying Party are materially prejudiced by such failure to give notice. The indemnifying Party shall notify in English the indemnified Party of its intentions as to the defense of the Claim or potential Claim in writing within ten (10) Business Days after receipt of notice of the Claim. If the indemnifying Party assumes the defense of a Claim against an indemnified Party, the indemnifying Party shall have no obligation or liability under this Section 13 as to any Claim for which settlement or compromise of such Claim or an offer of settlement or compromise of such Claim is made by an indemnified Party without the prior written consent of the indemnifying Party, which consent shall not be unreasonably withheld, conditioned or delayed. |
| (b) | Control of Defense. The indemnifying Party shall assume exclusive control of the defense and settlement (including all decisions relating to litigation, defense and appeal) of any such Claim (so long as it has confirmed its indemnification obligation responsibility to such indemnified Party under this Section 13.3 with respect to a given Claim); provided, however, that the indemnifying Party may not settle such Claim in any manner that would require payment by the indemnified Party, or would materially adversely affect the rights granted to the indemnified Party hereunder, or would materially conflict with the terms of this Agreement, without first obtaining the indemnified Party’s prior written consent, which consent shall not be unreasonably withheld. |
| (c) | Obligation to Cooperate. The indemnified Party shall reasonably cooperate with the indemnifying Party in its defense of the Claim including, without limitation, making relevant documents and records reasonably available for review and copying and making relevant persons within its control available for pertinent testimony at reasonable times and dates in accordance with the confidentiality provisions of Section 11 at the indemnifying Party’s expense. If the indemnifying Party assumes defense of the Claim, an indemnified Party may participate in, but not control, the defense of such Claim using attorneys of its choice and at its sole cost and expense. If an indemnifying Party does not agree to assume the defense of the Claim asserted against the indemnified Party (or does not give notice that it is assuming such defense), or if the indemnifying Party assumes the defense of the Claim in accordance with this Section 13.3 yet fails to defend or take other reasonable, timely action, in response to such Claim asserted against the indemnified Party, the indemnified Party shall have the right to defend or take other reasonable action to defend its interests in such proceedings, and shall have the right to litigate, settle or otherwise dispose of any such Claim and the indemnifying Party shall be obligated to indemnify the indemnified Party for any amounts expended by the indemnified Party; provided, however, that no Party shall have the right to settle a Claim in a manner that would adversely affect the rights granted to the other Party hereunder, or would materially conflict with this Agreement, or would require a payment by the Party, or adversely affect the Party or its Affiliates or its products outside the Territory, without the prior written consent of the Party entitled to control the defense of such Claim, which consent shall not be unreasonably withheld. |
| 40 |
| 14. | Term and Termination |
| 14.1 | Term |
This terms of this Agreement shall enter into force on the Effective Date, with the exception of Sections 7.1(a), 11, 16.3, 16.4, 16.5, 16.6, 16.7, 16.9, 16.10 and 16.14 which shall enter into force on the Signing Date, and shall remain in force until the expiration of the Royalty Term if not terminated earlier in accordance with this Agreement (“Term”). Upon expiration of the Royalty Term, and if HARROW has made all payments due to NOVALIQ as of such date, the license granted under Section 2.1 under: (i) the NOVALIQ Know-How shall become non-exclusive, fully paid-up, perpetual, and irrevocable; (ii) upon the request and at the cost of HARROW, NOVALIQ shall assign and transfer to HARROW the VEVYE Trademarks; and (iii) HARROW shall be entitled to continue to manage the www.veyve.com domain.
| 14.2 | Termination for Convenience |
At any time after the payment of the Second Upfront Payment, HARROW shall have the right to terminate this Agreement by providing six (6) months prior written notice to NOVALIQ.
| 14.3 | Termination for Cause |
In the event that either Party (“Breaching Party”) commits a material breach or default of any of its obligations hereunder, the other Party hereto (“Non-Breaching Party”) may give the Breaching Party written notice of such material breach or default, and shall request that such material breach or default be cured as soon as reasonably practicable. In the event that the Breaching Party fails to cure such breach or default within sixty (60) calendar days after the date of the Non-Breaching Party’s written notice thereof (in the event of default of payment within thirty (30) calendar days after the date of the Non-Breaching Party’s notice), the Non-Breaching Party may terminate this Agreement by giving written notice of termination to the Breaching Party. In the event the Breaching Party indicates in writing that it will be unable or is unwilling to cure the breach, this Agreement may be terminated by the Non-Breaching Party with immediate effect. Termination of this Agreement in accordance with this Section 14.3 shall not affect or impair the Non-Breaching Party’s right to pursue any legal remedy, including the right to recover damages, for any harm suffered or incurred by the Non-Breaching Party as a result of such breach or default.
| 14.4 | Termination for Challenge of NOVALIQ Patent Rights |
NOVALIQ may terminate this Agreement upon giving fourteen (14) days’ written notice to HARROW if HARROW or any of its Affiliates (directly or indirectly, e.g. by supporting a Third Party, individually or in association with any other person or entity) challenges the validity of the NOVALIQ Patent Rights or the corresponding Patent Rights owned by NOVALIQ outside the Territory in a legal proceeding before a court of competent jurisdiction. Any such termination shall only become effective if HARROW, its Affiliate or the Third Party supported by HARROW or any of its Affiliates, as the case may be, has not withdrawn such action before the end of the notice period. If a Sublicensee of HARROW challenges the validity of a NOVALIQ Patent Right or a corresponding Patent Right owed by NOVALIQ outside the Territory, NOVALIQ may terminate this Agreement upon giving thirty (30) days’ written notice to HARROW. Any such termination shall only become effective if the Sublicensee has not withdrawn such action and HARROW has not terminated the respective sublicense agreement before the end of the notice period.
| 41 |
| 14.5 | Termination for Safety Reasons. |
HARROW shall have the right to terminate this Agreement at any time upon providing thirty (30) days prior written notice to NOVALIQ: (a) if senior executives responsible for HARROW’s pharmacovigilance and clinical science functions determine in good faith after consultation with, and receipt of a written communication from a Regulatory Authority that the risk/benefit profile of the Licensed Product is such that the Licensed Product cannot continue to be Developed or Marketed or administered to patients safely; or (b) upon the occurrence of serious adverse events related to the use of the Licensed Product that after consultation with, and receipt of a written communication from a Regulatory Authority cause HARROW to conclude that the continued use of the Licensed Product by patients will result in patients being exposed to a product in which the risks outweigh the benefits. During the thirty (30) day notice period, the Parties shall begin to wind-down their respective activities under the Agreement to the extent related to the Licensed Product.
| 15. | Consequences of termination |
| 15.1 | Termination Due to NOVALIQ’s Breach |
If HARROW terminates this Agreement under Section 14.3, as of the effective date of such termination the following shall apply:
| (a) | No later than sixty (60) calendar days after the effective date of such termination, each Party shall return or cause to be returned to the other Party or, at the other Party’s option, destroy (and certify in writing the destruction of) all Confidential Information in tangible form received from the other Party and all copies in any medium thereof; provided, however, that each Party may retain any Confidential Information reasonably necessary for such Party’s continued practice under any license(s) which do not terminate pursuant to this Section 15.1, and may retain the Confidential Information solely for the purpose of ensuring its compliance with this Agreement and Applicable Laws by electronic files created in the ordinary course of business during automatic system back-up procedures pursuant to its electronic record retention and destruction practices that apply to its own general electronic files and information so long as such electronic files are (A) maintained only on centralized storage servers (and not on personal computers or devices), (B) not accessible by any of its personnel (other than its information technology specialists), and (C) are not otherwise accessed subsequently except with the written consent of the other Party or as required by law. Such retained copies of documents and Confidential Information shall remain subject to the confidentiality and non-use obligations set forth in this Agreement. |
| (b) | Each Party shall pay all amounts then due and owing as of the termination effective date. |
| (c) | Effective only in the event of such termination, NOVALIQ hereby grants to HARROW an exclusive (even as to NOVALIQ) and sublicensable license in the Field and in the Territory under the NOVALIQ Technology to Develop, Manufacture and Market the Licensed Product, provided, however, that any payment obligations under Section 7 shall survive the termination of the Agreement in consideration for the exclusive license of HARROW under this Section 15.1(c). |
| (d) | Except as set forth in this Section 15.1 and for the surviving provisions set forth in Section 15.3, the rights and obligations of the Parties hereunder shall terminate. |
| 42 |
For the avoidance of doubt, the rights and obligations of HARROW and NOVALIQ under this Section 15.1 in the event of a termination due to NOVALIQ’s material breach shall not exceed or be in any way broader than the rights and obligation of HARROW and NOVALIQ under the Agreement.
| 15.2 | Termination Due to HARROW’s Breach, HARROW’s Challenge of a NOVALIQ Patent Right or HARROW’s Termination under Section 14.2 or Section 14.5 |
If NOVALIQ terminates this Agreement under Section 14.3 or Section 14.4 or if HARROW terminates this Agreement under Section 14.2 or Section 14.5, as of the effective date of such termination the following shall apply:
| (a) | HARROW’s license under Section 2 of this Agreement automatically lapses and all rights to the NOVALIQ Technology automatically revert back to NOVALIQ. |
| (b) | No later than sixty (60) calendar days after the effective date of such termination, each Party shall return or cause to be returned to the other Party or, at the other Party’s option, destroy (and certify in writing the destruction of), all Confidential Information of the Disclosing Party in tangible form received from the other Party and all copies in any medium thereof; provided, however, that each Party may retain any Confidential Information reasonably necessary for such Party’s continued practice under any license(s) which do not terminate pursuant to this Section 14.2, and may retain the Confidential Information solely for the purpose of ensuring its compliance with this Agreement and Applicable Laws by electronic files created in the ordinary course of business during automatic system back-up procedures pursuant to its electronic record retention and destruction practices that apply to its own general electronic files and information so long as such electronic files are (A) maintained only on centralized storage servers (and not on personal computers or devices), (B) not accessible by any of its personnel (other than its information technology specialists), and (C) are not otherwise accessed subsequently except with the written consent of the other Party or as required by law. Such retained copies of documents and Confidential Information shall remain subject to the confidentiality and non-use obligations set forth in this Agreement. |
| (c) | Each Party shall pay all amounts then due and owing as of the termination effective date. |
| (d) | HARROW shall assign to NOVALIQ all of its right, title and interest in and to HARROW’s share in Joint Know-How and Joint Patent Rights and any and all rights, title and interest in and to Improvements (including Know-How and Patent Rights). |
| (e) | HARROW agrees to grant, and hereby grants to NOVALIQ, a cost-free exclusive license under the HARROW Technology to the extent required by NOVALIQ for the Development, Manufacture or Marketing of the Licensed Product in the Field in the Territory. |
| (f) | NOVALIQ shall have the right to request in writing within thirty (30) calendar days after the effective date of such termination, at HARROW’s cost and expense: |
| (i) | a complete copy of all Development Data and Regulatory Data to be provided in original form (i.e., with the relevant signatures and as suitable for submission for Regulatory Authorities) and access to all other Know-How in HARROW’s possession or under its Control relating to the Licensed Product, such Development Data and other Know-How to be provided within thirty (30) calendar days after the request; and |
| 43 |
| (ii) | the transfer of Regulatory Approvals, Reimbursement and Pricing Approvals held by HARROW, its Affiliates and/or Sublicensees (to the extent such transfer is possible), and if such Regulatory Approvals have not been obtained by HARROW, its Affiliates and/or Sublicensees as of the date of termination, HARROW shall inform NOVALIQ of the status of any such Regulatory Approvals or filings in progress and provide to NOVALIQ any such Licensed Product Regulatory Approvals in progress prepared by HARROW, its Affiliates and/or Sublicensees, provided, however, that such applications are provided on an “AS IS” basis. |
| (g) | Upon the request of NOVALIQ and to the extent permitted by Applicable Law, HARROW shall assign to NOVALIQ all distribution agreements and contracts with, or grants by, insurers or any licenses held by HARROW that are required for the Marketing of the Licensed Product for the Territory. |
| (h) | NOVALIQ shall have the right to assume activities under Sections 10.3(b) and 10.4(b) with respect to NOVALIQ Patent Rights and Joint Patent Rights. |
| (i) | HARROW and its Affiliates, Sublicensees shall be entitled, during the six (6) month period immediately following the effective date of termination, to finish any work-in-progress and to sell the Licensed Products remaining in inventory, in accordance with the terms of this Agreement; provided, however, that NOVALIQ shall have the right to purchase from HARROW any Licensed Products remaining in inventory as of the effective date of termination (that are not then committed to be supplied to any Third Party in the ordinary course of business), at a price equal to HARROW’s cost of goods. |
| (j) | HARROW shall transfer management responsibilities for the www.veyve.com domain back to NOVALIQ’s web hosting provider within five (5) Business Days of receipt of such request from NOVALIQ. |
| (k) | Except as set forth in this Section 15.2, in Section 15.1 and for the surviving provisions set forth in Section 15.3, the rights and obligations of the Parties hereunder shall terminate. |
| 15.3 | Effect of Expiration or Termination; Survival |
Expiration or termination of this Agreement shall not relieve the Parties of any obligation accruing prior to such expiration or termination. Any expiration or termination of this Agreement shall be without prejudice to the rights of either Party against the other accrued or accruing under this Agreement prior to expiration or termination. The provisions of Article 1, Section 3.7(b), Sections 7.2 and 7.4 (with respect to sales of Licensed Product made prior to termination or expiration), Section 7.5, Article 11, Section 12.6, Section 12.7, Article 13, Section 14.1 (second sentence), Article 15, Section 16.3, Section 16.4 and Section 16.5 shall survive the expiration or termination of this Agreement.
| 16. | General Provisions |
| 16.1 | Assignment |
This Agreement may not be assigned or otherwise transferred by either Party without the prior written consent of the other Party, which consent will not be unreasonably withheld, conditioned or delayed; provided, however, each of the Parties may, without such consent, assign this Agreement and its rights and obligations hereunder to any of its Affiliates or in connection with the transfer or sale of all or substantially all of the portion of its business to which this Agreement relates. Any permitted assignee will assume all obligations of its assignor under this Agreement in writing concurrent with the assignment. Any purported assignment in violation of this Section 16.1 will be void. NOVALIQ shall be entitled to assign its rights and claims under this Agreement to any Affiliate and any Third Party without HARROW’s consent.
| 44 |
| 16.2 | Force Majeure |
If the performance of any part of this Agreement by either Party, or any obligation under this Agreement, is prevented, restricted, interfered with or delayed by reason of any cause beyond the reasonable control of the Party liable to perform, the Party so affected shall, upon giving written notice to the other Party, be excused from such performance to the extent of such prevention, restriction, interference or delay, provided that the affected Party shall use its Diligent Efforts to avoid or remove such causes of non-performance and shall continue performance with the utmost dispatch whenever such causes are removed. When such circumstances arise, the Parties shall discuss what, if any, modification of the terms of this Agreement may be required in order to arrive at an equitable solution.
| 16.3 | Notices and Financial Accounting Communication |
All notices which are required or permitted hereunder shall be in writing and sufficient if delivered personally, sent by e-mail (and promptly confirmed by personal delivery, registered or certified mail or overnight courier), sent by nationally recognized overnight courier or sent by registered or certified mail, postage prepaid, return receipt requested, addressed as follows:
| (a) | if to NOVALIQ, addressed to: |
Novaliq
GmbH
Im Neuenheimer Feld 515
69120 Heidelberg
Germany
Email: c
Attention: CEO
With copy to:
Haley Lemon
| (b) | if to HARROW, addressed to: |
Harrow Health, Inc.
Harrow Eye, LLC
Harrow
IP, LLC
102 Woodmont Blvd.,
Suite 610, Nashville,
United States of America
TN 37205
Email: Attention: Chief Financial Officer
With copy to:
Polsinelli
PC
100 N. 4th Street
Suite 1000
St. Louis, MO 63102
Attn: Andrew M. Solomon
Email:
| 45 |
or to such other address(es) as the Party to whom notice is to be given may have furnished to the other Party in writing in accordance herewith. Any such notice shall be deemed to have been given: (a) when delivered if personally delivered or sent by facsimile on a Business Day (or if delivered or sent on a non-business day, then on the next Business Day); (b) on the Business Day after dispatch if sent by nationally-recognized overnight courier; or (c) on the fifth (5th) Business Day following the date of mailing, if sent by mail.
Any communication relating to financial accounting/accounts receivables (such as but not limited to invoices, credit notes and status of supplier account) shall be directed as follows:
If to NOVALIQ:
If to HARROW:
| 16.4 | Governing Law |
This Agreement and all disputes arising hereunder, shall be exclusively governed by, and interpreted and enforced in accordance with the laws of the State of New York, USA, without reference to any choice or conflict of law rules that would cause the application of laws of any other jurisdiction, except that questions affecting the construction and effect of any NOVALIQ Patent Rights shall be determined by the law of the country in which the NOVALIQ Patent Rights shall have been granted.
| 16.5 | Dispute Resolution |
| (a) | In the event of any dispute arising out of or in connection with this Agreement that cannot be settled by good faith negotiations over a period of thirty (30) calendar days within the JSC, and thereafter over an additional period of thirty (30) calendar days between senior management representatives of the Parties, the Parties agree to try to solve such dispute amicably by mediation. The Parties shall conduct a mediation procedure according to the Mediation Rules of the International Chamber of Commerce (“ICC”) in effect on the date of the commencement of the mediation proceedings. The location of the mediation proceedings will be New York City. The number of mediators will be one (1). The language of the mediation proceeding will be English. If the dispute has not been settled pursuant to the said rules within sixty (60) calendar days following the filing of a request for mediation or within such other period as the Parties may agree in writing, either Party may submit the dispute to final and binding arbitration. |
| (b) | Any dispute relating to the validity, performance, construction or interpretation of this Agreement, which cannot be resolved amicably between the Parties after following the procedure set forth in Section 16.5(a), shall be submitted to arbitration under the rules of the ICC, which rules deem to be incorporated by reference into this section. The arbitration shall be conducted by one (1) arbitrator selected by the Parties. Failing such agreement, the arbitrator shall be appointed by the ICC. Such arbitration shall be conducted in the English language and shall be held in Heidelberg, Germany if brought by HARROW and New York City, United States, if brought by NOVALIQ. The determination of the arbitration shall be final, binding and conclusive upon the Parties hereto, and enforceable in any court having jurisdiction. Each Party will bear its own costs and expenses (including its attorney’s fees) associated with any arbitration initiated under this section, and each Party will bear an equal share of the arbitrators’ and administrative fees associated with any arbitration initiated under this section. Notwithstanding the provisions of this Section 16.5(b), each Party shall have the right to seek preliminary or permanent injunctive relief in any court of competent jurisdiction as such Party deems necessary to preserve its rights and to protect its interests. |
| 46 |
| 16.6 | Severability |
If any provision of this Agreement is determined by any court or administrative tribunal of competent jurisdiction to be invalid or unenforceable, the Parties shall negotiate in good faith a replacement provision that is commercially equivalent, to the maximum extent permitted by Applicable Laws, to such invalid or unenforceable provision. The invalidity or unenforceability of any provision of this Agreement shall not affect the validity or enforceability of the other provisions of this Agreement.
| 16.7 | Entire Agreement and Amendments |
This Agreement, together with the Confidentiality Agreement and all Exhibits attached hereto, constitutes the entire agreement between the Parties regarding the Licensed Product, and supersedes all prior agreements, understandings and communications between the Parties, with respect to the Licensed Product. No modification or amendment of this Agreement shall be binding upon the Parties unless in writing and executed by the duly authorized representative of each of the Parties; this shall also apply to any change of this clause.
| 16.8 | Waivers |
The failure by either Party hereto to assert any of its rights hereunder, including the right to terminate this Agreement due to a breach or default by the other Party hereto, shall not be deemed to constitute a waiver by that Party of its right thereafter to enforce each and every provision of this Agreement in accordance with its terms.
| 16.9 | Counterparts |
This Agreement may be executed in any number of counterparts, each of which shall be deemed an original but all of which together shall constitute one and the same instrument.
| 16.10 | Independent Contractors |
The Parties are independent contractors and this Agreement shall not constitute or give rise to an employer-employee, agency, partnership or joint venture relationship among the Parties and each Party’s performance hereunder is that of a separate, independent entity.
| 16.11 | Language |
This Agreement, and any amendments or modifications thereto, shall be executed in the English language. No translation, if any, of this Agreement into any other language shall be of any force or effect in the interpretation of this Agreement or in determination of the intent of either of the Parties hereto.
| 16.12 | Headings |
The headings are placed herein merely as a matter of convenience and shall not affect the construction or interpretation of any of the provisions of this Agreement.
| 16.13 | Third Parties |
None of the provisions of this Agreement shall be for the benefit of or enforceable by any Third Party which shall be a Third Party beneficiary to this Agreement.
| 16.14 | Costs |
Except as is otherwise expressly set forth herein, each Party shall bear its own expenses in connection with the activities contemplated and performed hereunder.
- Signature pages follow -
| 47 |
IN WITNESS WHEREOF, this Agreement has been signed by the Parties hereto in two (2) originals, each Party acknowledging receipt of one original.
| NOVALIQ GMBH | ||
| By: | /s/ Dr. Christian Roesky | |
| Name: | Dr. Christian Roesky | |
| Title: | Chief Executive Officer | |
| By: | /s/ Dr. Oliver Schlüter | |
| Name: | Dr. Oliver Schlüter | |
| Title: | Chief Financial Officer |
IN WITNESS WHEREOF, this Agreement has been signed by the Parties hereto in two (2) originals, each Party acknowledging receipt of one original.
HARROW HEALTH, INC. HARROW IP, LLC |
||
| By: | /s/ Mark L. Baum | |
| Name: | Mark L. Baum | |
| Title: | Chief Executive Officer | |
HARROW EYE, LLC
| By: | /s/ Andrew R. Boll | |
| Name: | Andrew R. Boll | |
| Title: | Vice President |
Exhibit 10.29
EXECUTION VERSION
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
FIRST AMENDMENT TO LICENSE AGREEMENT
This First Amendment (the “First Amendment”) to the License Agreement dated June 6, 2023 (“Agreement”) is made and entered into as of the 14th day of February, 2024 (the “First Amendment Effective Date”) by and between NOVALIQ GMBH, a company organised under the laws of Germany and having its representative office at Im Neuenheimer Feld 515, 69120 Heidelberg, Germany (“NOVALIQ”); and HARROW, Inc. (formerly known as Harrow Health, Inc.) a company organised under the laws of Delaware and having its representative office at 102 Woodmont Blvd., Suite 610, Nashville, TN 37205, United States (“Harrow Health”), Harrow Eye, LLC, a limited liability company formed under the laws of Delaware and located at 102 Woodmont Blvd. Suite 610, Nashville, Tennessee 37205 United States (“Harrow Eye”) and Harrow IP, LLC, a limited liability company formed under the laws of Delaware and located at 102 Woodmont Blvd. Suite 610, Nashville, Tennessee 37205 United States (“Harrow IP”) (Harrow Health, Harrow Eye and Harrow IP being collectively referred to as “HARROW”). Novaliq and Harrow are sometimes referred to herein individually as a “Party” and collectively as the “Parties.”
WHEREAS, NOVALIQ and HARROW previously entered into the “Agreement”;
WHEREAS, HARROW wishes to sublicense certain rights under Section 2.1 of the Agreement to further Develop and Market the Licensed Product in the Field for the Canada Territory, as defined below; and
WHEREAS, NOVALIQ and HARROW desire to amend the Agreement as set forth herein.
NOW, THEREFORE, for good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, and with the specific intent to be bound hereby, the parties hereby agree as follows:
| 1. | DEFINITIONS AND INTERPRETATION |
| 1.1 | All capitalized terms used in this First Amendment but not defined shall have the meanings ascribed to them under the terms of the Agreement. |
| 1.2 | All references to “Harrow Health, Inc.” in the Agreement shall be interpreted to refer to Harrow, Inc. (formerly known as Harrow Health, Inc.). |
| 2. | AMENDMENTS |
| 2.1 | The following new sections shall be added to Article 1 of the Agreement: |
1.109 “Apotex” means Apotex Inc., a company incorporated under laws of Ontario with registered address at 150 Signet Drive, Toronto, Ontario, M9L 1T9.
1.110 “Apotex Sublicensing Agreement” shall have the meaning set forth in Section 2.2(c).
1.111 “Canada Territory” shall mean Canada, including its territories and possessions.
1.112 “United States Territory” shall mean the United States of America, including its territories and possessions
| Page 1 of 6 |
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
| 2.2 | A new Section 2.2(c) shall be included into the Agreement: |
“As of the First Amendment Effective Date, NOVALIQ expressly consents to the sublicensing of the rights granted to HARROW under Section 2.1 for the Canada Territory to Apotex in accordance with the terms and conditions of this Agreement (the “Apotex Sublicensing Agreement”). For the avoidance of doubt, Section 2.2(b) shall apply to the Apotex Sublicensing Agreement, and any further sublicensing by Apotex of the rights granted to Apotex under the Apotex Sublicensing Agreement shall be subject to Section 2.2(a). “
| 2.3 | Section 2.4(b) is amended and shall be replaced in its entirety by the following Section: |
“With the exception of Klarity Drops, Klarity C and Klarity C plus Loteprednol, during the Term, HARROW, its Affiliates or Sublicensees (excluding Apotex) holding rights to the Licensed Product in the Field in the Territory shall not develop or market, distribute, sell, offer to sell, or otherwise commercialize any prescription product for the treatment of DED in the Territory. In addition, HARROW, shall ensure that Apotex will not develop or market, distribute, sell, offer to sell, or otherwise commercialize any product for the treatment of DED or any directly related indication in the Canada Territory that contains semifluorinated alkanes. HARROW further agrees that it will not seek (and will procure that its Affiliates will not seek) to expand the product label indications in the United States Territory for any prescription product it markets and falls within one or more of the exceptions (a) – (e) as set forth in Section 1.21 to include the treatment of DED. Notwithstanding the above it shall not be a violation of this Section 2.4(b) for HARROW to maintain its passive minority investment interest in Surface Ophthalmics Inc.”
| 2.4 | Section 3.3(a) is amended and shall be replaced in its entirety by the following Section: |
“As between the Parties, HARROW shall be solely responsible for obtaining and maintaining all Regulatory Approvals for the Licensed Product in Canada and for interfacing, corresponding and meeting with Health Canada with respect to the Licensed Product, at its sole cost and expense. HARROW shall use Commercially Reasonable Efforts to file an NDA with Canada Health within two (2) years following the Effective Date. The Parties acknowledge and agree that HARROW may sublicense the rights and delegate the responsibilities and obligations under this Section 3.3(a) to Apotex under the Apotex Sublicense Agreement, provided that HARROW shall remain liable to NOVALIQ for any failure to comply with the obligations under this Section 3.3(a).”
| 2.5 | A new Section 5.5 shall be included into the Agreement: |
“5.5 Canadian Marketing of Vevye
As of the date that Health Canada approves the NDA for the Licensed Product, for the Canada Territory, HARROW shall ensure that VEVYE will be the primary focus in the promotion to ophthalmologists for their DED patients, and that VEVYE will occupy first position in such sales calls for DED. At the request of NOVALIQ, HARROW will provide copies of Apotex’s call activity reports against this target audience. Such reports shall be provided no more frequently than once per Calendar Quarter.”
| Page 2 of 6 |
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
| 2.6 | The title of Section 7.1 shall be replaced in its entirety by “Upfront Payments and Regulatory Milestone Payment” and a new Section 7.1(c) shall be included into the Agreement: |
“(c) a one-time regulatory milestone payment in the amount of [***] US Dollars in cash [***] within sixty-five (65) days of Apotex or any of its Affiliates obtaining the Marketing Authorization for the Licensed Product in the Canada Territory. HARROW shall notify NOVALIQ of the achievement of such regulatory milestone event within ten (10) Business Days upon its achievement. The regulatory milestone payment shall be made by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW); invoicing will be made by way of a credit note.”
| 2.7 | Section 7.4(a) is amended and shall be replaced in its entirety by the following Section: |
“(a) Royalty Payments. During the Royalty Term HARROW shall pay to NOVALIQ royalties, calculated on the basis of aggregated annual Net Sales of Licensed Product in the United States Territory as follows:
| Royalty Tiers | Royalty Rate | |
| For the portion of aggregated Net Sales of Licensed Product in the United States Territory that is lower than [***] | [***] | |
| For the portion of aggregated Net Sales of Licensed Product in the United States Territory that is equal to or [***] | [***] | |
| For the portion of aggregated Net Sales of Licensed Product in the United States Territory that is equal to or exceeding [***] | [***] |
| 2.8 | Section 7.4(e) is amended and shall be replaced in its entirety by the following Section (e) and a new Section 7.4(f) shall be included into the Agreement: |
“(e) During the Term, HARROW shall pay to NOVALIQ [***] of all royalty payments received from any sublicensee for the Canada Territory (“Sublicense Income”).
(f) Reports; Payment of Royalties and Sublicense Income. HARROW shall report to NOVALIQ the date of the First Commercial Sale of the Licensed Product in the United States Territory and the Canada Territory within ten (10) Business Days of its respective occurrence. In addition, during the term of this Agreement following the First Commercial Sale of the Licensed Product in the Territory, HARROW shall furnish to NOVALIQ a written report for each Calendar Quarter showing (on a country-by-country basis) the united sold of Licensed Products, the gross sales of Licensed Products, the calculation of Net Sales (including the deductions from gross sales) of the Licensed Product by HARROW, its Affiliates and Sublicensees in the Territory during the reporting period, and, for the United States Territory, the royalties payable under this Agreement, and for the Canada Territory, the royalties payable to HARROW under the Apotex Sublicense Agreement and the Sublicense Income payable under this Agreement. Reports shall be due within forty-five (45) calendar days after the end of each Calendar Quarter. At least once a year, HARROW’s independent auditors shall review royalty reports, calculations and associated payments to NOVALIQ. If any prior period adjustments are required by HARROW or its independent auditors to the Net Sales calculation, such adjustments shall be clearly noted in and applied to the following period royalty calculation, report and payment (e.g., the next quarterly report). Royalty payments and Sublicense Income payments shall be made by HARROW by wire transfer to NOVALIQ’s bank account (as notified by NOVALIQ to HARROW from time to time), with any transfer fees to be paid by HARROW, in immediately available funds within five (5) Business Days after delivery of a final quarterly royalty report by way of a credit note.”
| Page 3 of 6 |
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
| 2.9 | Section 10.6 is amended and shall be replaced in its entirety by the following Section: |
“HARROW shall use the NOVALIQ Trademarks (i.e., VEVYE and EYESOL), as needed, in the United States to mark the Licensed Product. NOVALIQ shall have the sole right to file, prosecute, maintain, defend and enforce the NOVALIQ Trademarks in the Territory. HARROW shall procure that Apotex will use the VEVYE Trademark to mark the Licensed Product in the Canada Territory.”
| 2.10 | Section 11.6 is amended and shall be replaced in its entirety by the following Section: |
“The Parties agree that the material terms of this Agreement are the Confidential Information of both Parties, subject to the special authorized disclosure provisions set forth in Sections 11.2 and 11.3 and this Section 11.6. The Parties have agreed to make a joint press release of the execution of this Agreement and will agree on the wording of such joint press release following the Effective Date. After the release of such joint press release, no disclosure of the existence, or the terms, of this Agreement may be made by either Party without first obtaining the prior written approval of the other Party and agreement upon the nature and text of such public announcement and no Party shall use the name, trademark, trade name or logo of the other Party, its Affiliates or their respective employee(s) in any publicity, promotion, news release or disclosure relating to this Agreement or its subject matter, without the prior express written permission of the other Party, except as may be required by Applicable Law or as otherwise expressly provided in this Agreement. Notwithstanding the aforementioned, NOVALIQ is entitled to use the name and logo of the Licensed Product and HARROW for public relation purposes in and outside of the Territory. The Parties agree that NOVALIQ shall be entitled to make a press release upon the achievement of each milestone and each milestone trigger of a royalty payment under this Agreement and HARROW shall refuse its permission to such press release only for cause and by suggesting amendments to the press release which would allow HARROW to grant its permission. No permission shall be required to the extent a Party is legally required (i) by Applicable Laws; (ii) by the listing standards or agreements of any national or international securities exchange or similar laws of a governmental authority, market or agency; or (iii) in a judicial, administrative or arbitration proceedings, provided that in such instances the other Party shall be entitled to make a corresponding public announcement. Notwithstanding the above, as of the First Amendment Effective Date HARROW shall be entitled to issue a press release announcing that it has entered into the Apotex Sublicense Agreement to Develop and Market the Licensed Product in the Field for the Canada Territory.”
| 2.11 | A new Section 15.2(l) shall be included into the Agreement: |
“(l) In case of termination of this Agreement, NOVALIQ will negotiate with Apotex in good faith a direct license to Apotex for the Canada Territory on the basis of the terms of this Agreement and the Apotex Sublicense Agreement, except to the extent that Apotex is in material breach of this Agreement or the Apotex Sublicense Agreement (in which case, the Apotex Sublicense Agreement shall automatically terminate with the termination of this Agreement to the extent it relates to the Licensed Product for the Canada Territory, unless otherwise agreed between NOVALIQ and Apotex, at the sole discretion of NOVALIQ).
| 3. | In all other respects, the terms and conditions of the Agreement will remain in full force and effect as written; provided, however, that the terms and conditions of the original Agreement will prevail over the terms and conditions of this First Amendment to the extent there are any inconsistencies between this First Amendment and the Agreement, unless explicitly regulated otherwise. |
| Page 4 of 6 |
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
IN WITNESS WHEREOF, the Parties have executed this First Amendment as of the First Amendment Effective Date in two (2) originals, each Party acknowledging receipt of one original.
NOVALIQ GmbH
| By: | /s/ Christian Roesky | |
| Name: | Christian Roesky | |
| Title: | CEO/ Managing Director |
| By: | /s/ Oliver Schluter | |
| Name: | Oliver Schluter | |
| Title: | CFO |
| Page 5 of 6 |
Certain identified information has been excluded from this exhibit because it is both (i) not material and (ii) is the type that the registrant treats as private or confidential and would likely cause competitive harm to the registrant if publicly disclosed. [***] indicates omitted information.
IN WITNESS WHEREOF, the Parties have executed this First Amendment as of the First Amendment Effective Date in two (2) originals, each Party acknowledging receipt of one original.
Harrow, INc.
| By: | /s/ Mark L. Baum | |
| Name: | Mark L. Baum | |
| Title: | CEO |
Harrow IP LLC
| By: | /s/ Mark L. Baum | |
| Name: | Mark L. Baum | |
| Title: | CEO |
Harrow EYE, LLC
| By | /s/ Andrew R. Boll | |
| Name: | Andrew R. Boll | |
| Title: | Vice President |
| Page 6 of 6 |
Exhibit 10.30
Consulting Agreement
THIS CONSULTING AGREEMENT (the “Agreement”) is effective on March 1, 2026, and is entered into by and between John Saharek with a principal address located at (“Consultant”) and Harrow, Inc., a Delaware corporation with its principal address located at 1A Burton Hills Blvd., Suite 200, Nashville, Tennessee (the “Company”).
WHEREAS, the Company wishes to retain Consultant as an advisor to the Company; and
WHEREAS, Consultant wishes to provide advisory services to the Company as set forth below.
NOW THEREFORE, in consideration of the mutual promises contained herein and other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, Consultant and the Company agree, intending to be legally bound, as follows:
| 1. | Consulting Services. |
| 1.1. | Consultant will provide consulting services to the Company during the Term of this Agreement. The consulting services (“Services”) are set forth in the Statement of Work (“SOW”) that is attached hereto as Appendix A and made a part hereof, as it may be amended from time to time by the parties hereto. |
| 1.2. | Consultant represents and warrants to the Company that: (a) Consultant has the required skill, experience and qualifications to perform the Services, shall perform the Services in a professional and workmanlike manner in accordance with generally recognized industry standards for similar services and shall devote sufficient resources to ensure that the Services are performed in a timely and reliable manner; and (b) Consultant shall perform the Services in compliance with all applicable federal, state and local laws and regulations. |
| 2. | Effective Date; Term and Termination. |
| 2.1. | This Agreement shall be effective on the later of the dates that it is executed by the Company and Consultant (the “Effective Date”) and shall terminate upon the date Services are completed (the “Term”) unless: (i) this Agreement is sooner terminated as provided in Section 2.2 below; or (ii) the parties agree in writing to extend the Term for a mutually agreed upon period. |
| 2.2. | The Agreement and the Services provided by Consultant may be terminated by Consultant or the Company, at any time and for any reason, upon five (5) days prior written notice of termination. |
| 2.3. | Consultant shall immediately cease to provide any further Services under this Agreement and/or any applicable SOW upon receipt of notice from the Company that the Company is terminating this Agreement and/or such SOW. Upon termination of this Agreement, Consultant shall be entitled to payment for Services completed prior to such termination. Thereafter, the Company shall owe Consultant no further amounts or obligations. |
| 3. | Consulting Fees. |
| 3.1. | In consideration of the Services provided hereunder, the Company shall pay Consultant consulting fees as set forth in the applicable SOW (“Consulting Fee”). |
| 3.2. | Consultant shall be responsible for all expenses incurred in association with performance of the Services, unless pre-approved by the Company in writing in advance. |
| 4. | Ownership of Intellectual Property / Inventions / Work Product. Consultant agrees that Consultant shall disclose promptly to the Company all inventions, ideas, concepts, and discoveries, including but not limited to processes, methods, formulas, biological materials, specimens, chemical compounds, formulations, software, data, techniques, products, applications, systems, procedures, technical information, drawings, reports and designs as well as improvements and modifications thereof and know-how thereto (whether or not protectable by copyright, patent, trademark, trade secret or any other proprietary rights), that Consultant makes, conceives of, discovers or develops as a result of providing the Services or that arise from the Confidential Information defined in Section 6 below, as well as the Deliverables required under the SOW (collectively “Work Product”). Consultant agrees that all Work Product shall be the sole and exclusive property of the Company. |
| 4.1. | Consultant represents and warrants that all Work Product is and shall be Consultant’s original work (except for material in the public domain or provided by the Company) and, to the best of Consultant’s knowledge, does not and will not violate or infringe upon the intellectual property right or any other right whatsoever of any person, firm, corporation or other entity. |
| 4.2. | Consultant agrees that any Work Product, if subject to copyright, shall be considered a “work made for hire” within the meaning of the Copyright Act of 1976, as amended (the “Act”). If and to the extent that any Work Product is found as a matter of law not to be a “work made for hire” within the meaning of the Act, Consultant agrees to assign, and by this Agreement and Consultant’s signature below, Consultant hereby does assign to the Company all right, title and interest in and to Work Product, and all copies thereof, and the copyright, patent, trademark, trade secret and all other proprietary rights in Work Product. |
| 4.3. | Consultant agrees that, at the request of the Company, Consultant will execute all such documents and perform all such acts as the Company or its duly authorized agents may reasonably require: (a) to effect the assignment of Work Product as agreed above; (b) to apply for, obtain, and vest in the name of the Company alone patents, patent applications, copyrights or other intellectual property rights in any country and (c) at the Company’s expense, to assist the Company in prosecuting any such rights. |
| 4.4. | Consultant agrees that promptly upon termination of this Agreement, Consultant shall deliver to the Company all Work Product, either completed or uncompleted, and any documents, reports and other materials which are in Consultant’s possession in connection with the performance of Services under this Agreement. |
| 2 |
| 4.5. | To the extent, if any, that Consultant retains any right, title or interest in or to any Work Product, Consultant hereby grants to Company a perpetual, irrevocable, fully paid-up, transferable, sublicensable, exclusive, worldwide right and license (a) to reproduce, distribute, display and perform (whether publicly or otherwise), prepare derivative works of and otherwise modify, make, use, sell, offer to sell, import and otherwise exploit (and have others exercise such rights on behalf of Company) all or any portion of such Work Product, in any form or media (now known or later developed); (b) to modify all or any portion of such Work Product, including the making of additions to or deletions from such Work Product, regardless of the medium (now or hereafter known) into which such Work Product may be modified and regardless of the effect of such modifications on the integrity of such Work Product; (c) to identify Consultant, or not to identify Consultant, as one or more authors of or contributors to such Work Product or any portion thereof, whether or not such Work Product or any portion thereof have been modified; and (d) to otherwise exploit such Work Product in any manner whatsoever; in each case without notice to, the consent of, or accounting to Consultant. Consultant further waives any “moral” rights or other rights with respect to attribution of authorship or integrity of such Work Product that Consultant may have under any applicable law, whether under copyright, trademark, unfair competition, defamation, right of privacy, contract, tort or other legal theory. |
| 5. | Confidentiality. Consultant acknowledges that Consultant will receive confidential and proprietary information from, on behalf of, or at the direction of, the Company in connection with, and during the course of providing, the Services, including but not limited to technical, clinical, marketing, commercial and/or legal information, data, reports, drawings, models, designs, prototypes, biological material, specimens, chemical compounds, formulas, manufacturing or other processes, software, specifications, patent applications, marketing strategies, customer information and customer lists (“Confidential Information”). As used herein, “Confidential Information” includes Work Product as defined in Section 5. All Confidential Information is and shall at all times remain the exclusive property of the Company. Consultant agrees: |
| 5.1. | to hold the Confidential Information in strict confidence and not to disclose or make available any Confidential Information to any third party whatsoever, without the prior written consent of the Company; |
| 5.2. | to use the Confidential Information only for the benefit of the Company and only for the purpose of providing the Services; |
| 5.3. | to take at least the same degree of care to prevent disclosure of Confidential Information as Consultant takes to preserve and safeguard Consultant’s own confidential and proprietary information, but in any event, no less than a reasonable degree of care; |
| 5.4. | not to make copies of the Confidential Information except to the extent that the copies are reasonably necessary for providing the Services; |
| 3 |
| 5.5. | to return or destroy (as the Company may direct) any Confidential Information held by Consultant immediately upon termination of the Term of this Agreement pursuant to Section 2 above and provide the Company with a letter certifying that all such Confidential Information has been returned or destroyed as directed; |
| 5.6. | that Confidential Information excludes information that: (a) as evidenced by Consultant’s written records, was lawfully known to Consultant prior to its communication by, on behalf of, or at the direction of the Company and was not communicated to Consultant subject to any restrictions on disclosure or use; or (b) as evidenced by Consultant’s written records, is independently developed by Consultant without use or knowledge of the Confidential Information; or (c) is or becomes a part of the public domain other than by a breach of this Agreement by Consultant; (d) becomes known to Consultant by the action of a third party not in breach of any obligation of confidence; or (e) is required to be disclosed or made available by Consultant to a third party pursuant to any applicable law, governmental regulation, or decision of any court or tribunal of competent jurisdiction, so long as Consultant takes reasonable steps, in light of the circumstances, to give the Company sufficient prior notice in order to contest such law, governmental regulation, or decision; |
| 5.7. | that no representation or warranty, express or implied, is made by the Company as to the accuracy, completeness or reasonableness of any Confidential Information and that neither the Company will have any liability to Consultant as a result of Consultant’s possession or use of the Confidential information; and |
| 5.8. | that money damages may not be sufficient remedy for any breach of this Section 6 and that the Company will be entitled to seek specific performance and injunctive or equitable relief as a remedy for any such breach. |
| 5.9. | Nothing in this Section 6 is intended to limit any remedy of the Company under Tennessee Uniform Trade Secrets Act, or otherwise available under law. |
| 5.10. | Consultant shall, at Harrow’s request and in Harrow’s sole discretion, execute separate Confidentiality Agreements and, if given access to patient health information, a “Business Associate Agreements” as required by the U.S. Health Insurance Portability and Accountability Act of 1996 (“HIPAA”). |
| 6. | Conflicts of Interest. |
| 6.1. | Consultant represents and warrants that Consultant is not under any pre-existing obligation in conflict or in any way inconsistent with the provisions of this Agreement. Consultant represents and warrants that Consultant’s performance of all the terms of this Agreement will not (a) breach any agreement to keep in confidence proprietary information acquired by Consultant in confidence or in trust prior to commencement of this Agreement, or (b) breach any other agreement with any third party. Consultant warrants that Consultant has the right to disclose and/or or use all ideas, processes, techniques and other information, if any, which Consultant has gained from third parties, and which Consultant discloses to the Company or uses in the course of performance of this Agreement, without liability to such third parties. Notwithstanding the foregoing, Consultant agrees that Consultant shall not bundle with or incorporate into any deliveries provided to the Company herewith any third party products, ideas, processes, or other techniques, without the express, written prior approval of the Company. Consultant represents and warrants that Consultant has not granted and will not grant any rights or licenses to any intellectual property or technology that would conflict with Consultant’s obligations under this Agreement. Consultant will not knowingly infringe upon any copyright, patent, trade secret or other property right of any former client, employer or third party in the performance of the Services. |
| 4 |
| 6.2. | The Company acknowledges that Consultant may perform consulting services for other clients. Consultant represents and warrants that as of the Effective Date, there is no conflict of interest which would prevent Consultant from performing the Services for the Company, and that Consultant is not under any legal or contractual relationship with any third party which is inconsistent with any provision of this Agreement. During the Term of this Agreement, Consultant will use Consultant’s best efforts not to enter into any other agreement or arrangement that will directly compete with the Services to be rendered hereunder. In the event that Consultant becomes aware of any potential or actual conflicts of interest regarding the provision of the Services, Consultant shall promptly disclose the fact and nature of such conflict to the Company. Thereafter, upon the Company’s request, Consultant shall cease to provide further Services to the Company, or to be entitled to receive any further compensation in consideration of the provision of further Services, to the extent specified in writing by the Company. |
| 7. | Debarred Person. Consultant hereby certifies that Consultant is not currently nor has been debarred by the U.S. Food and Drug Administration pursuant to 21 USC §335a(a) or (b), or under any similar law or regulation by the European Medicines Evaluation Agency or any other national or regulatory authority or agency. If Consultant becomes aware that Consultant is or becomes the subject of any debarment or similar proceedings in any jurisdiction, then Consultant shall promptly notify the Company. |
| 5 |
| 8. | Independent Contractor. The relationship of Consultant to the Company shall be that of an independent contractor rendering professional services. Consultant is not an employee of the Company. Nothing contained in this Agreement shall be deemed to create a relationship of employer and employee or principal and agent between the Company and Consultant. In no circumstance shall Consultant look to the Company as Consultant’s employer, partner, agent or principal. In further demonstration of Consultant’s independent consultant status, Consultant and the Company agree as follows: |
| 8.1. | No Benefits, Etc. Consultant shall have no authority to execute contracts or make commitments on behalf of the Company or assume any obligation or liability on behalf of the Company or to negotiate with third parties regarding any matters relating to the Company and its business. Consultant has the right to perform services for others during the term of this Agreement subject to any limitations and/or restrictive covenants that predate this Agreement (all of which, for the avoidance of doubt, are unaffected by this Agreement). Consultant shall not receive any training from the Company in the skills necessary to perform the Services required by this Agreement. The Company shall not require Consultant to devote full time to performing the Services required by this Agreement. Consultant shall not be entitled to participate in any of the benefit, welfare, bonus or incentive plans maintained by the Company for its employees. Consultant’s exclusion from benefit programs maintained by the Company is a material component of the terms of compensation negotiated by the parties including, without limitation, worker’s compensation insurance, travel accident insurance, medical/dental insurance, life insurance, short- term and/or state disability insurance or benefits, long-term disability insurance, holiday pay, sick pay, paid vacation, bonuses, salary continuation pay, leaves of absence (paid or unpaid), pension plan benefits, retirement savings plan benefits or lease vehicle benefits. To the extent that Consultant may become eligible for any benefit programs maintained by the Company, Consultant hereby waives Consultant’s right to participate in such programs. The Company shall not make any state or federal unemployment compensation payments on behalf of Consultant, and Consultant shall not be entitled to these benefits in connection with Services performed under this Agreement. Consultant will not apply for any government-sponsored benefits that are intended to apply to employees, including, but not limited to, state and federal unemployment benefits. Consultant is solely responsible for all insurance applicable to Consultant, including, without limitation, worker’s compensation insurance, travel accident insurance, medical/dental insurance, life insurance, short- term and/or state disability insurance or benefits, and long-term disability insurance. |
| 8.2. | Taxes & Expenses. Consultant shall be responsible for all tax payments due from him in accordance with federal, state, city, county and other local tax laws, including all applicable income taxes. Under no circumstances will the Company: (a) withhold FICA (Social Security and Medicare taxes) from Consultant’s payments or make FICA payments on Consultant’s behalf; or (b) withhold state or federal income taxes from Consultant’s payments. Consultant shall be responsible for all expenses incurred while performing Services under this Agreement, including without limitation (i) insurance premiums (ii) license fees and all expense incurred with respect to license fees; and (iii) all memberships and dues. The Company will reimburse Consultant for pre-approved travel-related expenses incurred by Consultant directly in connection with the Services, as set forth more fully in Section 4 above. |
| 9. | Tax Indemnity. Consultant agrees to indemnify and hold harmless the Company from any and all claims or demands under the Internal Revenue Code of 1986, as amended, or any state or local tax law or ordinance in respect of any failure of the Company to withhold income tax, FICA or any other tax from the Consulting Fees paid to Consultant, including any interest or penalties relating thereto and any costs or expenses incurred in defending such claims. |
| 10. | Assignment. Consultant shall not assign this Agreement or any of its rights or privileges without the prior written consent of the Company, which consent the Company may grant or withhold in its sole discretion. The Company may assign this Agreement to any party that agrees to assume this Agreement and all of the Company’s duties and obligations thereunder. |
| 11. | Waiver. No waiver of this Agreement or any of its provisions shall be binding upon a party unless in writing and signed by each party. The waiver by either party of a breach or violation of any provision of this Agreement shall not constitute or be construed as a waiver of any subsequent breach or violation of that provision or as a waiver of any breach or violation of any other provision. |
| 6 |
| 12. | Severability. If any provision of this Agreement is held by a court of competent jurisdiction to be invalid, illegal or unenforceable, the remaining provisions of this Agreement shall be unimpaired, and the invalid, illegal or unenforceable provision shall be replaced by a mutually acceptable provision, which, being valid, legal and enforceable, comes closest to the intention of the parties underlying the invalid, illegal or unenforceable provision. |
| 13. | Survival. The provisions of Sections 2.3, 5, 6, 9-19 and any other obligation under this Agreement which is to survive or be performed after termination of this Agreement, regardless of the cause therefor, shall survive any termination or expiration of this Agreement. |
| 14. | Notices. Any notice or other communication required or permitted to be made or given under this Agreement to either party shall be in writing and shall be sufficiently given if (i) hand delivered, (ii) sent by overnight guaranteed delivery service, such as Federal Express or UPS; or (iii) sent by facsimile transmission or electronic mail during addressee’s normal business hours, with a duplicate copy sent by overnight delivery or certified or registered mail (except for any notice of termination which must be sent by method (i) or (ii)), addressed as follows: |
| If to Consultant: | John Saharek | ||
| If to the Company: | Harrow, Inc. | ||
| 1A Burton Hills Blvd. Suite 200 | |||
| Nashville, TN 37215 | |||
| Attn: Mark Baum |
or to such other address or addressee as either party may from time to time designate to the other by written notice. Any such notice or other communication shall be deemed to be given as of the date it is received by the addressee.
| 15. | Publicity. Consultant shall not make any public announcements in respect of this Agreement without the prior written consent of the Company. |
| 16. | Advice of Counsel. Each party acknowledges that, in executing this Agreement, such party has had the opportunity to seek the advice of independent legal counsel and has read and understood all of the terms and provisions of this Agreement. This Agreement shall not be construed against any party by reason of the drafting or preparation hereof. |
| 17. | Governing Law. This Agreement shall be governed by and construed in accordance with the laws of the State of Tennessee, excluding the choice of law rules, and the parties hereby agree to submit to the jurisdiction and venue of the State and Federal courts of the State of Tennessee, and agree that the State and Federal courts of the State of Tennessee shall be the exclusive forum for the resolution of all disputes related to or arising out of this Agreement. |
| 18. | Entire Agreement; Amendments. This Agreement, including any applicable SOW, represents the entire agreement between the parties in relation to the subject matter contained herein and supersedes all previous other agreements and representations, whether oral or written. This Agreement may be modified only if such modification is in writing and signed by a duly authorized representative of each party. |
***SIGNATURE PAGE FOLLOWS***
| 7 |
SIGNATURE PAGE
IN WITNESS WHEREOF, the parties hereto have executed this Consulting Agreement as of the date first above written.
| CONSULTANT: | ||
| By: | /s/ John Saharek | |
| Name: | John Saharek | |
| An individual | ||
| Date: | February 27, 2026 | |
| COMPANY: | ||
| HARROW, INC. | ||
| By: | /s/ Mark L. Baum | |
| Name: | Mark L. Baum | |
| Title: | Chairman & CEO | |
| Date: | February 27, 2026 | |
| 8 |
Appendix A
Statement of Work
under Consulting Agreement
by and between
John Saharek and Harrow, Inc.
Effective Date: March 1, 2026 (or the Agreement Effective Date, if later)
Program: IHEEZO Anterior Clinic Adoption Pilot (“Program”)
Pilot Duration: SOW term begins on Effective Date and will continue through the term date December 31, 2026, unless earlier terminated per the Agreement.
Background and Objective
The Company desires to launch and evaluate a focused commercial initiative (“IHEEZO Anterior Adoption Pilot”) designed to drive sustainable, clinic-based adoption of IHEEZO for office-based ophthalmic procedures. The objective of the Program is to protect and expand existing clinic utilization, generate durable clinic revenue, and produce a scalable commercial, reimbursement, and execution playbook for broader deployment over a 12-month pilot period.
Consultant Role
Consultant will serve as Program Lead, providing overall strategy, governance, targeting discipline, execution oversight, and senior-level engagement support (including private equity platform engagement as applicable).
Fees and Payment
Consulting fees and any performance-based components will be as set forth below:
Base Consulting Fee: $18,540 per every two (2) weeks.
Annual Performance/Incentive Component based on adoption of IHEEZO
| 9 |
Exhibit 21.1
HARROW,
INC. SUBSIDIARIES
as of December 31, 2025
Name of Subsidiary |
|
State of Incorporation or Organization |
ImprimisRx, LLC |
|
Delaware |
Imprimis NJOF, LLC |
|
New Jersey |
ImprimisRx NJ, LLC |
|
New Jersey |
Harrow Eye, LLC |
|
Delaware |
Harrow IP, LLC |
|
Delaware |
ImprimisRx Nashville, LLC |
|
Delaware |
Harrow Analytical Services, LLC Melt Pharmaceuticals Inc. |
|
Delaware Delaware |
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in Registration Statement Nos. 333-265244 and 333-215672 on Form S-3 and Registration Statement Nos. 333-288167, 333-257413, 333-220186, 333-198674, 333-183488, and 333-159159 on Form S-8 of our reports dated March 2, 2026, relating to the financial statements of Harrow, Inc. and the effectiveness of Harrow, Inc.’s internal control over financial reporting appearing in this Annual Report on Form 10-K for the year ended December 31, 2025.
| /s/ Deloitte & Touche LLP | |
| Nashville, Tennessee | |
| March 2, 2026 |
Exhibit 23.2
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in Registration Statements on Form S-8 (Nos. 333-288167, 333-257413, 333-220186, 333-198674, 333-183488, and 333-159159) and Form S-3 (No. 333-215672) of Harrow, Inc. of our reported dated March 27, 2025 on the consolidated balance sheet of Harrow, Inc. as of December 31, 2024 and the related consolidated statements of operations, stockholders’ equity, and cash flows for the year ended December 31, 2024, appearing in this Annual Report on Form 10-K of Harrow, Inc. for the year ended December 31, 2025.
| /s/ Crowe LLP |
Costa Mesa, California
March 2, 2026
Exhibit 23.3
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in Registration Statement Nos. 333-288167, 333-257413, 333-220186, 333-198674, 333-183488, and 333-159159 on Form S-8 and Registration Statement No. 333-215672 on Form S-3 of our report dated March 19, 2024 (March 27, 2025 as to Note 19), relating to the consolidated statements of operations, stockholders’ equity, and cash flows for the year ended December 31, 2023 of Harrow, Inc., appearing in this Annual Report on Form 10-K of Harrow, Inc. for the year ended December 31, 2025.
/s/ KMJ Corbin & Company LLP
Glendora,
California
March 2, 2026
Exhibit 31.1
CERTIFICATION OF THE PRINCIPAL EXECUTIVE OFFICER UNDER
SECTION 302 OF THE SARBANES-OXLEY ACT
I, Mark L. Baum, certify that:
| (1) | I have reviewed this Form 10-K for the fiscal year ended December 31, 2025 of Harrow, Inc.; | |
| (2) | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; | |
| (3) | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; | |
| (4) | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| d) | Disclosed in the report any change in this registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| (5) | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: March 2, 2026 | /s/ Mark L. Baum |
| Mark L. Baum | |
| Chief Executive Officer |
EXHIBIT 31.2
CERTIFICATION OF THE PRINCIPAL FINANCIAL OFFICER UNDER
SECTION 302 OF THE SARBANES-OXLEY ACT
I, Andrew R. Boll, certify that:
| (1) | I have reviewed this Form 10-K for the fiscal year ended December 31, 2025 of Harrow, Inc.; | |
| (2) | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; | |
| (3) | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; | |
| (4) | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| d) | Disclosed in the report any change in this registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| (5) | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: March 2, 2026 | /s/ Andrew R. Boll |
| Andrew R. Boll | |
| President and Chief Financial Officer |
Exhibit 32.1
HARROW, INC.
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Mark L. Baum, Chief Executive Officer of Harrow Inc. (the “Company”), do hereby certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that to the best of my knowledge:
(1) the Annual Report on Form 10-K of the Company for the annual period ended December 31, 2025 (the “Report”) fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
| Date: March 2, 2026 | |
| /s/ Mark L. Baum | |
| Mark L. Baum | |
| Chief Executive Officer |
The foregoing certification is being furnished as an exhibit to the Report pursuant to Item 601(b)(32) of Regulation S-K and Section 1350 of Title 18 of the United States Code and, accordingly, is not being filed with the U.S. Securities and Exchange Commission as part of the Report and is not to be incorporated by reference into any filing of the Company under the Securities Act of 1933 or the Securities Exchange Act of 1934 (whether made before or after the date of the Report, irrespective of any general incorporation language contained in such filing).
Exhibit 32.2
HARROW, INC.
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Andrew R. Boll, Chief Financial Officer of Harrow Inc. (the “Company”), do hereby certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that to the best of my knowledge:
(1) the Annual Report on Form 10-K of the Company for the annual period ended December 31, 2025 (the “Report”) fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: March 2, 2026 |
|
|
|
/s/ Andrew R. Boll |
|
Andrew R. Boll |
|
President and Chief Financial Officer |
|
The foregoing certification is being furnished as an exhibit to the Report pursuant to Item 601(b)(32) of Regulation S-K and Section 1350 of Title 18 of the United States Code and, accordingly, is not being filed with the U.S. Securities and Exchange Commission as part of the Report and is not to be incorporated by reference into any filing of the Company under the Securities Act of 1933 or the Securities Exchange Act of 1934 (whether made before or after the date of the Report, irrespective of any general incorporation language contained in such filing).